Una
neonata con ipotonia ed un viso particolare
1UO
terapia Intensiva Pediatrica e Neonatale Osp. M.bufalini Cesena
2UO
Genetica Medica Area Vasta Romagna
3Pediatra
di Famiglia
Indirizzo
per corrispondenza:
abiasini@ausl-cesena.emr.it
An
hypotonic newborn with a special look
Key
words
Congenital
hypotonia, Joint-meeting, Communication
Abstract
The
cure and care of the children with congenital chronic
disabilities is the real challenge for the hospital-territorial
network cooperative model. This organizational strategy will
achieve success only if all the caregivers work strictly together
to obtain the best assistance of the children and their families.
A case report of a newborn with global muscles weakness and a
particular face, is described outlining this way of caring. The
reader is driven to the correct diagnosis by following a logical
approach. Adequate communication of the disease is of crucial
importance and was made during the joint-meeting between the
healthcare team and the family. This is the appropriate method to
maintain a good relationship, giving parents hope despite the
awareness of the uncertainty of outcomes. |
R.R. è
una bimba nata a termine (38 sett + 1 gg) da parto indotto per
polidramnios (circa 7 l di liquido amniotico allagano la sala parto
al momento della rottura delle membrane), che esita in taglio cesareo
urgente per decelerazioni al tracciato cardiotocografico. Alla
nascita la neonata si presenta ipotonica con una respirazione
insufficiente, la frequenza cardiaca (FC) è > 100 battiti
per minuto (bpm); viene assistita con ventilazione non invasiva in
maschera e ossigeno (FiO2 max 0.9) fino alla comparsa di
respiri superficiali ma efficaci. Si assiste al miglioramento della
ossigenazione che alla pulsossimetria (SaO2) non supera
l86-90%. Viene quindi trasferita alla nostra struttura con
ossigeno (FiO2 0.30) in culla (APGAR 5 a 1,8 a 5;
peso 3110 (>50°), lunghezza 52 cm (50°), circonferenza
cranica 35,8 cm (<97°). Allesame obiettivo la facies
appare sui generis con ipotonia marcata e amimia: la bocca è
mantenuta aperta e le labbra hanno una caratteristica disposizione a
tenda; non vi sono altre note dismorfiche se non una lieve
prominenza frontale che simula la presenza di bozze e una
micrognatia. I piedi sono entrambi torti tipo equino varo supinato, è
presente un edema cutaneo diffuso, i genitali esterni sono nella
norma, lipotonia senza movimenti spontanei (general
movements) è più evidente al collo, la postura è
a rana con gli arti inferiori flessi sul tronco, la
suzione è scarsa o assente, i riflessi arcaici sono evocabili,
presenti quelli osteotendinei (ROT), lobiettività
cardiaca è nella norma.
Cosa
diciamo ai genitori? Cerchiamo di non far male parlando di possibile
malattia già presente alla nascita che si dovrà
precisare con esami e tempo, osservando la capacità della
piccola di respirare bene in modo autonomo e di nutrirsi da sola; i
genitori faticano a capire ma non chiedono di più
concentrandosi sullassistenza alla bambina.
Nelle
prime 48 ore di vita R.R. ha richiesto solo un minimo fabbisogno di
ossigeno, lipotonia iniziale è progressivamente
diminuita con comparsa di deboli movimenti agli arti superiori e
collo, mentre quelli inferiori sono rimasti fermi, mantenendo
liniziale postura a rana, la bocca, per gran parte del tempo
aperta, ha continuato a mostrare la particolare forma delle labbra a
tenda; ledema diffuso si è progressivamente
riassorbito. La suzione compare e migliora progressivamente sensibile
alla intensa stimolazione della madre e degli operatori
(fisioterapiste/infermiere), con buona coordinazione
suzione/deglutizione/respirazione. La madre è sempre stata
positivamente impegnata verso la sua bimba col canto di filastrocche
e la lettura del libro in reparto. Nessuna anomalia alle indagini
laboratoristiche e strumentali (ecografia cerebrale, addominale,
cardiaca, Elettrocardiogramma (ECG), radiografia del torace, Fundus
Oculi, Otoemissioni Acustiche).
Il
percorso diagnostico si profila nel capitolo delle ipotonie
neonatali ricercando lobiettività più
specifica di ogni patologia. Micrognazia, piede torto, edema diffuso
sono segni secondari alla scarsa motilità fetale del terzo
trimestre e quindi non utili a inquadrare le cause del difetto.
Ancora una volta è la storia familiare, raccolta in corso di
consulenza genetica, che guida alla diagnosi corretta. Sono infatti
emersi tre casi di familiari consanguinei per linea materna che hanno
sviluppato cataratta giovanile. Questo elemento, in relazione alla
sintomatogia presentata è già patognomonico. Inoltre la
madre della piccola presenta lievissimi, ma inequivocabili segni
della malattia: pur non presentando i segni della miotonia alle mani,
la facies si presenta amimica con il labbro inferiore leggermente
protruso, il viso ha un ovale tipicamente allungato con il mento
appuntito; in aggiunta, pur essendo assente la ptosi palpebrale,
riferisce però un certo grado di affaticabilità dopo
sforzo serale comparso relativamente di recente. Questa ricorrenza è
molto specifica della Distrofia miotonica di Steinert (DMS) e
il sospetto diagnostico è coerente con laspetto clinico
della bimba1. La DMS è confermata dallindagine
genetica (analisi molecolare che ha mostrato amplificazione delle
triplette CTG nella regione 3 UTR non tradotta del gene DMPK
in un range >150-1000 triplette), il cariotipo è risultato
normale. Lentità numerica di amplificazione delle
triplette correla con lepoca di esordio clinico, attualmente è
in corso la valutazione genetica sulla madre.
Una
diagnosi differenziale ragionata e semplice
L'ipotonia
neonatale, in assenza di note dismorfiche che richiamino un quadro
sindromico complesso, e in assenza di chiari segni dallarme
prenatale a eziopatogenesi definita (distacco di placenta, prolasso
di funicolo o sua rottura) che possano far pensare a una Sindrome
Ipossico Ischemica, deve essere messa in relazione con il sospetto
di2:
- Distrofia miotonica tipo 1 (DMS) (discriminabile a livello molecolare dal riscontro di mutazioni nel gene DMPK )
- Distrofia miotonica tipo 2 (DM2) (discriminabile a livello molecolare dal riscontro di mutazioni nel gene CNBP )
- Atrofia muscolare spinale (SMA) (discriminabile a livello molecolare dal riscontro di mutazioni nel gene SMN1) (i ROT sono assenti)
Se la
lunghezza del frammento CGT del gene DMPK è nel range di
normalità e se l'analisi molecolare del gene CNBP ed
eventualmente del gene SMN1 ha dato esito nella norma occorre
valutare la possibilità di effettuare una valutazione EMG ed
eventualmente una biopsia di muscolo per avviare la diagnosi
differenziale verso le miopatie congenite che sono diagnosticabili a
livello molecolare. La presenza di note dismorfiche tipiche guiderà
peraltro alla sindrome specifica (es. sindrome di Down, Prader Willi
(PWS) o DMS ).
In questo
caso la presentazione era tale da orientare fortemente il sospetto
diagnostico (Figura 1):
- la presenza dei ROT e lassenza di fascicolazioni linguali ha allontanato la SMA1.
- la normalità dei genitali ha allontanato la PWS
- la familiarità era assolutamente tipica per DMS
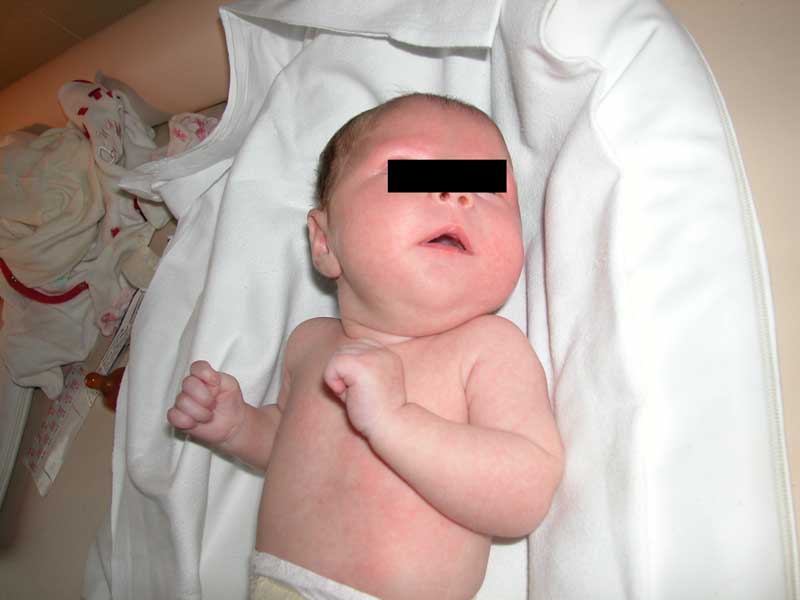
La
malattia di Steinert ha una prevalenza di 1/20.000. Si associa
a mutazioni di un locus sul cromosoma 19q13-2 (ripetizione anomala
della tripletta CTG, ovvero > 34 ripetizioni, con maggiore
penetranza allelica se > 50 ripetizioni)3. La
trasmissione è autosomica dominante e può verificarsi
anticipazione (ossia la malattia tende sempre di più
ad aggravarsi e manifestarsi più precocemente nelle
generazioni successive). La diagnosi è confermata dalla
genetica molecolare che rileva le anomalie del gene localizzato su
19q13-2. Tre forme sono descritte4 : La forma lieve: è
presente solo la cataratta giovanile, una lieve miotonia o il diabete
mellito. La forma classica colpisce diversi organi: la sintomatologia
predominante è la debolezza dei muscoli distali e facciali con
ptosi palpebrale, miotonia agli arti che impedisce le normali
attività; a livello cardiaco sono frequenti i difetti di
conduzione (90%) che possono portare a morte e spesso si associano
alla morte in culla, a livello intestinale possono presentare
disfagia, alterazioni dellalvo con diarrea e stipsi fino alla
pseudo ostruzione, aumento delle transaminasi; sono presenti deficit
intellettivi disturbi psicotici di tipo ossessivo-compulsivi,
ipersomnia ed apnee nel sonno, cataratta oculare; coesistono disturbi
endocrini: iperinsulinismo, diabete, ipogonadismo con infertilità,
deficit di GH; tipica è la calvizie frontale. La forma
congenita è causata dalla ripetizione di un gran numero di
triplette CTG che per lo più deriva dal ramo materno: maggiore
è il numero di triplette più grave è la
sintomatologia clinica. Spesso cè insufficienza
respiratoria, i deficit intellettivi con atrofia cerebrale sono
presenti nel 50-60%, è possibile lautismo e la
deambulazione ha un atteggiamento miopatico. In presenza di familiari
affetti è possibile effettuare diagnosi prenatale con prelievo
di villi coriali a circa 12 settimane di età gestazionale ed
estrazione del DNA: la diagnosi è definita dalla presenza di
una espansione dellallele DMPK ma non è tuttora
possibile predire se il feto con mutazione avrà la forma
congenita o una distrofia miotonica di un altro tipo (Tabella
I).
Segni
e Sintomi |
Forma
Lieve |
Forma
Classica |
Forma
Congenita |
Miotonia
Cataratta
giovanile |
Si
Si |
Si
Si |
Si |
Debolezza
muscoli distali,facciali,bocca a tenda
Aritmie
cardiache
Disfagia,stipsi,diarrea
Calvizie
frontale |
Si
Si
Si
Si
Si |
Si
Si | |
Insufficienza
respirat.
Malform.
Cong. arti
Atrofia
cerebrale
Autismo
/ Psicosi |
Si |
Si
Si
Si
Si |
Discutiamo
di tutto ciò col pediatra di famiglia della piccola, il
genetista, la psicologa, la coordinatrice e linfermiera che
seguì R.R. durante il ricovero e prepariamo il joint-meeting
(JM) cioè lincontro con i genitori per la comunicazione
della diagnosi e la discussione dei problemi che R.R. si troverà
ad affrontare. Ci ripetiamo che limpatto relazionale sarà
più condizionato dal come diremo le cose e meno
dalle parole che verranno dimenticate, che latmosfera di
empatia che sapremo creare, pur nella chiarezza espositiva della
malattia, farà sentire i genitori meno perduti limitando le
fantasie catastrofiche.
Adotteremo,
per lincontro, il protocollo SPIKES (Setting, Perception,
Invitation, Knowledge, Empaty, Strategy)5 che consiste nei
seguenti passi: a) preparare il contesto pronti allascolto; b)
esplorare ciò che i genitori sanno già; c) dare solo le
informazioni importanti per i genitori; d) fornire alla coppia i dati
necessarie a comprendere la clinica; e) lasciare emergere le
emozioni; f) concordare il piano di cura; il pdf potrà
accogliere i bisogni fisici, emozionali, psicologici e spirituali
della famiglia se sarà valorizzato e sostenuto dagli
ospedalieri. R.R. ha una malattia rara ed averla identificata in modo
preciso è comunque positivo per la famiglia. In genere il 40%
dei pazienti con una malattia rara ha ricevuto una diagnosi errata
prima di quella corretta6. Lassistenza di cui R.R.
avrà bisogno viene definita come media e
permanente.
Con
queste premesse, il JM famiglia-operatori avvenne in modo
soddisfacente ed efficace a circa un mese di vita: erano presenti i
genitori con la piccola, il neonatologo, il genetista e linfermiera
dedicata a R.R. mentre il pdf partecipò in viva voce
telefonica. La comunicazione ufficiale della diagnosi, con la
consegna alla famiglia della descrizione molecolare della DMS avvenne
durante lincontro; i genitori, durante il periodo trascorso a
casa, avevano avuto modo di rinforzare il loro attaccamento alla
piccola riconoscendone le risorse ed erano in parte preparati ad
accogliere la diagnosi.
Cosa
fare nel Follow up ?
La
gestione integrata dellassistenza alla piccola prevede
annualmente il controllo della glicemia e Hb Glicata, valutazione
della funzionalità tiroidea, degli ormoni sessuali e lECG/
Holter: lalta mortalità è data da complicazioni
cardiache. Va seguita la crescita e lo sviluppo neuroevolutivo,
prevedendo la fisioterapia respiratoria, kinesiterapia e la
correzione dei difetti ortopedici.
Ogni due
anni va eseguita la visita oculistica per cogliere la comparsa di
cataratta.
Vanno
evitati alcuni farmaci quali: fluidificanti, sedativi e morfina per
il rischio di depressione respiratoria e di occlusione intestinale,
antiartmici di classe A, Amiodarone e beta bloccanti per interferenza
sul ritmo, gli anticolinergici per ritenzione urinaria, le statine
per il possibile aumento della debolezza muscolare, e gli anestetici
per lipotetica ipertermia maligna e arresto respiratorio.
Attualmente
il pdf ha con la famiglia un buon rapporto e si sente telefonicamente
con gli ospedalieri che conducono il follow up alle cadenze previste;
racconta che R.R. ha ora 3 mesi, il peso è 4290 gr, la
lunghezza 56 cm , la CCr. 38,5 cm, succhia al seno e il pianto, prima
quasi impercettibile è diventato più forte e la piccola
si lascia ora consolare dalla madre. Linseguimento visivo e i
potenziali uditivi sono normali. Il tono muscolare è un pò
migliorato e i gessetti (Figura 2),
posti dopo intervento ortopedico, sono stati sostituiti con tutore a
permanenza (Figura 3). La relazione che
la madre ha con la figlia è soddisfacente.

Il caso
di R.R. può costituire un esempio di buona collaborazione fra
i vari specialisti dellospedale e territorio nellaffrontare
unitariamente una malattia cronica. Nello scenario attuale in cui ci
sono sempre meno bambini e meno malattie infettive, mentre i pediatri
lamentano la scarsità di problemi medici e la futilità
delle richieste delle famiglie, e aumenta linappropriato
accesso al Pronto Soccorso, dedicare risorse assistenziali e
relazionali alle malattie croniche e complesse che stanno aumentando,
è una saggezza culturale e professionale. Ci vuole coraggio da
ambedue le parti: gli ospedalieri devono cedere con ragionevolezza il
credito di fiducia che la famiglia ha loro affidato al team del
territorio (pdf e collaboratori) e questi devono essere allaltezza
del compito aumentando la loro disponibilità, cultura ed
empatia. Solo in questo modo la rete dimissione protetta
cesserà di essere appena uno schema, ben delineato da una
delibera regionale e attuato dalle aziende, ma spesso teorico e
quindi insufficiente nelle risposte, e divenire una vera rete di cura
efficace del bambino nella sua famiglia.
1.
Tonetto P, Spola R, Bagna R, et al. Steinerts
myotonic dystrophy: severe neonatal form with unknown family history.
Acta Biomed Ateneo Parmense 2000;71 Suppl 1:755-7.
2. Laugel
V,Cossee M, Matis J et al. Diagnostiv
approach to neonatal hypotonia: retrospective study on 144 neonate.
Eur J Pediatr. 2008 167(5). 517-23.
3. Bell,
D.B. Smith D.W. Myotonic dystrophy in the neonate.J Pediatr
1972;81(1):83-6.
4. Pearse
rR.G, Howeler CJ. Neonatal
form of Dystrophia myotonica: five cases in preterm babies and a
review of earlier reports. Arch Dis Child 1979;54(5):331-8.
5. Baile
WF, Buckman R, Lenzi R et al. SPIKES-A
six-step protocol for delivering bad news: application to the patient
with cancer. Oncologist 2000;5(4):302-11
6.
G.Tornese,V.Declich, G.Ciana et al Sintomi
comuni per malattie rare. Un approccio generale del pediatra ai
pazienti con malattie rare. Medico e Bambino 2007;26:230-36.