Quando
il dolore osseo viene... dal sangue
1Scuola
di Specializzazione in Pediatria, Ferrara; 2UOC di
Pediatria e Neonatologia, Azienda Ospedaliera di Ravenna; 3Pediatria
di Libera Scelta, AUSL di Ravenna; 4UO di Ortopedia e
Traumatologia Pediatrica, Istituto Ortopedico Rizzoli, Bologna
Indirizzo
per corrispondenza: lorenzo.mam@libero.it
When
bone pain comes from
the blood
Key
words Thalasso-drepanocytosis,
Mild anaemia, Osteonecrosis, Case report Abstract
The
paper describes the case of an 8-year-old Moroccan girl who
presented with recurrent multi-focal bone pains associated with
negative flogosis indexes and standard radiological exams. The
magnetic resonance exam of her thighbones showed a multi-focal
inflammation; bone biopsy showed that the inflammation was
aseptic and secondary to an osteonecrosis caused by
vaso-occlusive crisis. After the haemoglobin electrophoresis
performed in consideration of the persistent evidence of a mild
microcytic anaemia with anisocytosis, the diagnosis was of
thalasso-drepanocytosis.
Thalasso-drepanocytosis
is one of the sickle cell syndromes characterised by
heterozygosis associated with thalassemic haemoglobinopathy. It
can be clinically similar to homozygote drepanocytosis and
therefore it needs the same therapeutic approach and follow-up.
The
case highlights the importance of thinking about sickle cell
crisis, also in absence of important anaemization, when foreign
patients coming from worlds endemic areas for thalassemic
haemoglobinopathy present with recurrent bone pains. |
|
Presentiamo
il caso di una bambina di 8 anni di origine marocchina che giunge
alla nostra attenzione per la comparsa di dolori ossei ricorrenti
multifocali in apiressia associati a negatività degli indici
di flogosi e degli esami radiologici standard. Lesame RMN dei
due femori evidenzia un quadro infiammatorio multifocale che risulta
essere di carattere asettico alla biopsia ossea e che si rivela
secondario ad una osteonecrosi da crisi vasoocclusiva. La diagnosi è
di una talassodrepanocitosi condizione emersa allelettroforesi
emoglobinica eseguita in considerazione del riscontro persistente
(sia in corso di crisi che nei periodi intercritici) di una lieve
anemia microcitica (Hb media 11 g/dl) con anisocitosi allo striscio
periferico.
La
talassodrepanocitosi è una delle sindromi falcemizzanti
caratterizzate da una condizione di eterozigosi per il trait
falcemico associata ad unemoglobinopatia talassemica. Può
essere clinicamente affine ad una drepanocitosi omozigote e richiede
pertanto il medesimo approccio terapeutico e di follow-up.
Il caso
descritto evidenzia limportanza di pensare, in caso di dolori
ossei con carattere di ricorrenza in pazienti stranieri provenienti
da aree del mondo endemiche per le emoglobinopatie talassemiche, ad
una possibile causa da crisi falcemiche, anche in assenza di una
importante anemizzazione.
Y. è
una bambina marocchina di 8 anni che giunge alla nostra attenzione
per dolori ricorrenti da un anno localizzati alla porzione distale
della coscia sinistra, in alcuni episodi anche a carico della coscia
controlaterale e degli arti superiori; buona risposta alla terapia
antidolorifica e tipica risoluzione spontanea degli episodi in 2-3
giorni. Anamnesi familiare e patologica remota apparentemente
negative per elementi di rilievo (padre con diabete mellito
insulino-dipendente).
Negativa
la radiografia delle sedi interessate dal dolore (Figura
1a), agli esami ematici enzimi muscolari nella norma e
riscontro esclusivo di un incostante lieve rialzo degli indici di
flogosi (PCR max 33 mg/l). Allemocromo GB 6.650 (N 52%, L 30%,
M 11%), Hb 10,9 g/dl, MCV 63,5 fl, MCH 22,5 pg, RDW 15% e allo
striscio periferico riscontro di unanisocitosi con emazie a
bersaglio. Reticolociti 26 per mille.
Al
momento della nostra valutazione il dolore è di intensità
tale da compromettere la deambulazione e si associa ad un lieve
arrossamento della cute a livello dellestremità distale
della coscia sinistra; si riscontra una sostanziale negatività
degli indici di flogosi (PCR 8 mg/l, VES 5, fibrinogeno 410 mg/dl) e
lemocromo appare sovrapponibile a quello eseguito
precedentemente. Non epatosplenomegalia alla palpazione addominale.
Negative la radiografia del torace e lecografia addominale (in
particolare non evidenza di litiasi colecistica). Intradermoreazione
secondo Mantoux negativa.
Come
nelle altre occasioni lepisodio si autolimita in 48-72 ore ed
il dolore, di carattere multifocale, più intenso e frequente
allarto inferiore sinistro, sembra essere a partenza ossea.
Viene
quindi eseguita la RMN del bacino e di entrambi i femori che mostra
un quadro infiammatorio periostale e, bilateralmente, della
spongiosa, evidenti questi ultimi come aree disomogenee di elevato
segnale ad aspetto di strie longitudinali (Figura.
1b).
|
|
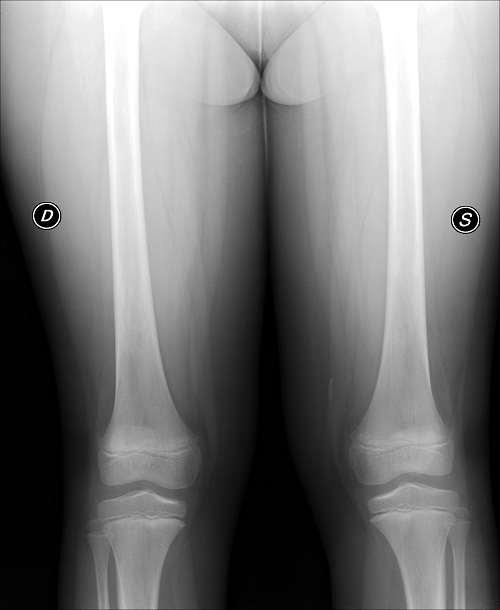
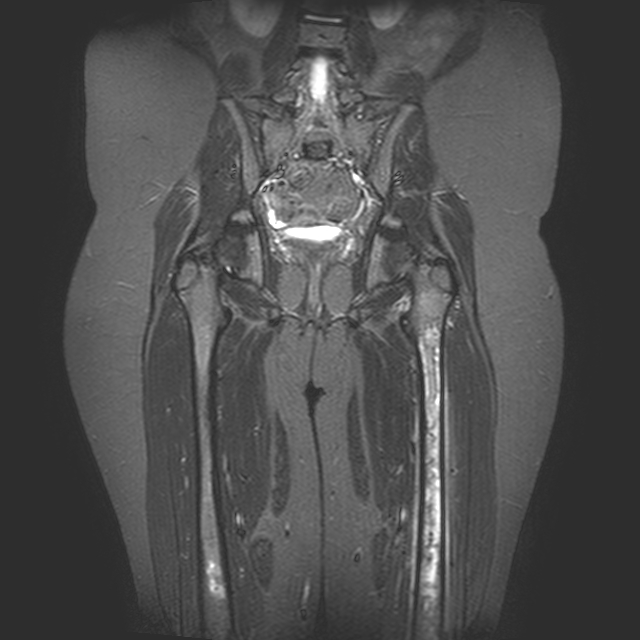
Figura
1.
a. Rx
femori eseguita in corso di crisi dolorosa ossea, negativa per
alterazioni della diafania di corticale e midollare.
b.
RMN femori eseguita durante periodo intercritico con evidenti aree
disomogenee di elevato segnale ad aspetto di strie longitudinali in
sede periostale e, anche a livello del femore destro (sede di
pregressi episodi dolorosi), della spongiosa
Alla
biopsia ossea si conferma un quadro di infiammazione cronica asettica
(escluse invece ipotetiche cause tumorali). In considerazione della
ricorrenza dei sintomi e del carattere migrante viene inizialmente
sospettata unosteomielite cronica multifocale ricorrente,
infiammazione ossea non batterica, idiopatica, a carico
prevalentemente delle metafisi delle ossa lunghe, che può
associarsi (anche se non frequentemente) ad una negatività di
indici di flogosi.
Sulla
base delle caratteristiche di imaging delle lesioni e del quadro
istologico viene però riconsiderato il dato della lieve anemia
microcitica, dellanisocitosi e dellaumento dei
reticolociti, per cui viene eseguita elettroforesi dellHb con
riscontro di un quadro di talassodrepanocitosi HbS/β+ (HbA2
5,6%, HbF 5,7%, HbS 62%) secondario allassociazione di un
trait falciforme paterno (HbA2 ed HbF normali, HbS 35%) e di un trait
βtalassemico materno (HbA2 5,5%, HbF 3,1%) (attualmente
in corso la ricerca delle mutazioni dei geni beta-globinici).
Viene
quindi impostata terapia con idrossiurea al dosaggio di 10/mg/kg/die
(con controlli periodici dellassetto emoglobinico ed
epato-renale).
Il quadro
osseo cronico ricorrente e multifocale presentato è pertanto
riconducibile ai foci di necrosi da crisi vaso-occlusive falcemiche.
La
talassodrepanocitosi (microdrepanocitosi, Sickle-Thal
nella letteratura anglosassone) è unemoglobinopatia
scoperta e descritta da Silvestroni e Bianco nel 1944 caratterizzata
dalla presenza contemporanea di un trait drepanocitico e di un trait
talassemico e quindi di un contemporaneo difetto qualitativo e
quantitativo dellemoglobina.
Frequente
soprattutto nellAfrica centro-occidentale, nel sud dellArabia
e in alcune zone di Italia, Spagna e Grecia (la prevalenza di
portatori del tratto drepanocitico nei 25 stati europei è di
1/150, in Africa centrale e occidentale è pari al 15-25%,
nelle Antille francesi al 10-15% e nell'area del Mediterraneo a
1-15%), è caratterizzata da un quadro clinico simile a quello
della drepanocitosi omozigote, sebbene possa essere più lieve
in caso di deficit non completo delle catene beta-globiniche. Il
quadro ematologico, in particolare, presenta tipicamente i caratteri
dellanemia microcitica con ridotto volume globulare medio,
ridotto contenuto emoglobinico ed alterazioni morfologiche delle
emazie affini a quelle riscontrabili nel semplice trait-talassemico,
sebbene in alcune condizioni possa anche osservarsi la caratteristica
deformazione a falce.
Il
decorso di questa e delle altre cosiddette sindromi
falcemizzanti, caratterizzate dalla eterozigosi composta di un
trait drepanocitico con un altro difetto qualitativo o quantitativo
della sintesi delle catene globiniche, appare però affine a
quello della malattia drepanocitica omozigote; una eccezione è
rappresentata però dai casi in cui si associa un trait
beta-talassemico lieve o laplotipo HbS Benin (aplotipo di HbS
associato ad un difetto falcizzante più lieve), entrambi
frequenti soprattutto in Sicilia.
Nella
nostra bambina non si sono riscontrate né una franca anemia,
anche in corso di crisi falcemiche vaso-occlusive, né
manifestazioni cliniche fino alletà di 7 anni; la
comparsa di crisi ricorrenti di osteonecrosi rappresenta però
ovviamente un chiaro carattere di gravità della condizione.
La
terapia in questi casi ricalca fondamentalmente quella prevista per
le anemie drepanocitiche: idrossiurea (induzione della produzione di
HbF) in caso di crisi dolorose ricorrenti (>2 allanno) o
sindromi toraciche ricorrenti severe, terapia trasfusionale cronica
(preferibile la procedura di exsanguinotrasfusione) prevalentemente
nella prevenzione dello stroke nei pazienti a rischio (alta velocità
di flusso al Doppler transcranico).
Nel Box 1
è riportato in sintesi lattuale protocollo di follow-up
e gestione terapeutica stilato dallAIEOP nel 2012 per i
bambini con anemia falciforme, anche per le forme di eterozigosi
composta S-β°.
Nel caso
della nostra paziente, nonostante lesordio tardivo e lanemia
lieve, è stata impostata terapia con idrossiurea al dosaggio
di 10/mg/kg/die (attualmente stabile, incrementabile fino a 35
mg/kg/die) in considerazione della ricorrenza delle crisi dolorose,.
Il caso
descritto vuole pertanto sottolineare come, in caso di crisi dolorose
ossee ricorrenti, soprattutto in un paziente originario delle
sopradette aree endemiche per il tratto drepanocitico, se è
presente una microcitemia anche in assenza di importante anemia sia
opportuna lesecuzione di una analisi dellemoglobina
mediante cromatografia HPLC, allo scopo di individuare la presenza di
eventuali varianti che possano causare falcizzazione.
|
Box
1. Sintesi delle Raccomandazioni per la gestione della
malattia drepanocitica in età pediatrica (AIEOP 2012) |
Bibliografia di riferimento
- Kliegman RM. Nelson Textbook of Pediatrics 19th ed. 2011:1663-70.
- Associazione Italiana Ematologia Oncologia Pediatrica (AIEOP), Gruppo di Lavoro Patologia del globulo rosso. Raccomandazioni per la gestione della malattia drepanocitica in età pediatrica in Italia. 2012. http://www.aieop.org/files/files_htmlarea/tutto%20giu12.pdf
- Bender MA, Hobbs W. Sickle Cell Disease. GeneReviews. Last revision 2012.
- Steinberg MH. Management of Sickle Cell Disease. N Engl J Med 1999;340:1021-30.
- Winfred C. Wang. Sickle Cell Anemia and other Sickling Syndromes. Wintrobe's Clinical Hematology, XII edition, Vol 1 (Greer J.P.). 2008:1038-82.
- Milner PF et al. Sickle cell disease as a cause of osteonecrosis of the femoral head. N Engl J Med 1991; 325:1476-81.
- Rees DC, Williams TN, Gladwin MT. Sickle cell disease. Lancet 2010;376(9757):2018-31.
- Orah S, Platt OS. Hydroxyurea for the treatment of sickle cell anemia. N Engl J Med. 2008;358(13):1362-9.
- Lazzerini M, Rabusin M. L'anemia falciforme. Medico e Bambino 2006;25(4):223-34.
Il
testo completo delle Raccomandazioni AIEOP è disponibile al
seguente indirizzo:
http://www.aieop.org/files/files_htmlarea/tutto%20giu12.pdf
Classificazione MeSH
Bibliografia
Kliegman RM. Nelson Textbook of Pediatrics 19th ed. 2011:1663-70.
Associazione Italiana Ematologia Oncologia Pediatrica (AIEOP), Gruppo di Lavoro Patologia del globulo rosso. Raccomandazioni per la gestione della malattia drepanocitica in età pediatrica in Italia. 2012. http://www.aieop.org/files/files_htmlarea/tutto%20giu12.pdf
Bender MA, Hobbs W. Sickle Cell Disease. GeneReviews. Last revision 2012.
Steinberg MH. Management of Sickle Cell Disease. N Engl J Med 1999;340:1021-30.
Winfred C. Wang. Sickle Cell Anemia and other Sickling Syndromes. Wintrobe's Clinical Hematology, XII edition, Vol 1 (Greer J.P.). 2008:1038-82.
Milner PF et al. Sickle cell disease as a cause of osteonecrosis of the femoral head. N Engl J Med 1991; 325:1476-81.
Rees DC, Williams TN, Gladwin MT. Sickle cell disease. Lancet 2010;376(9757):2018-31.
Orah S, Platt OS. Hydroxyurea for the treatment of sickle cell anemia. N Engl J Med. 2008;358(13):1362-9.
Lazzerini M, Rabusin M. L'anemia falciforme. Medico e Bambino 2006;25(4):223-34.