I meccanismi di azione dei Corticosteroidi
Indirizzo per la corrispondenza: bartolozzi@unifi.it
L'infiammazione
è una conseguenza dell'infezione: gli agenti infettivi che
superano le barriere epiteliali della cute e delle mucose attivano
direttamente i recettori del complemento e i recettori toll-like, i
due componenti principali del sistema immune innato. Il sistema
immune innato. insieme al sistema immune adattivo, costituisce un
elemento essenziale delle difese dell'individuo nei confronti
dell'infezione: esso rappresenta l'unico sistema di difesa nella
prima settimana dopo l'inizio dell'aggressione batterica, in
attesa che l'immunità adattiva (anticorpi e immunità
cellulare specifica) completi la sua formazione.
L'attivazione
della cascata del complemento e dei recettori toll-like porta alla
sintesi e alla liberazione dei mediatori dell'infiammazione che
hanno un'azione immediata sui capillari e sugli altri piccoli vasi:
la vasodilazione localizzata, l'aumentata permeabilità
cellulare, la fuoriuscita dai vasi delle proteine plasmatiche e la
migrazione dei leucociti nel tessuto colpito dall'infezione,
determinano i segni classici dell'infiammazione: calor, dolor,
rubor, tumor e functio laesa. Inizia così la produzione di
nuove citochine infiammatorie che attivano i leucociti, accorsi nel
tessuto interessato dall'infezione. I meccanismi omeostatici
anti-infiammatori contrastano con questi processi, contemporaneamente
all'eliminazione dell'agente infettivo da parte dell'immunità
innata e adattiva. L'asse ipotalamo-ipofisi surrene e in
particolare i glicocorticoidi sono essenziali nel limitare e
annullare il processo infiammatorio (Rhen T, Cidlowski JA.
Antiinfiammatory action of glucocorticoids - New mechanisms for old
drugs. N.Engl J Med 2005, 353:1711-23).
I glicocorticoidi,
insieme ai mineralocorticoidi, fanno parte dei corticosteroidi: essi
costituiscono le due classi di steroidi che vengono sintetizzate
fisiologicamente dalla corteccia surrenale. Di norma il
corticosurrene sintetizza ogni giorni 10 mg di idrocortisone
(cortisolo), di cui la maggior parte nelle ore del mattino o la parte
minore nel pomeriggio. Il cortisone, così diffuso in terapia
umana, non è sintetizzato dal corticosurrene, cioè esso
è un ormone, preparato in periferia a partire
dall'idrocortisone.
Nei meccanismi fisiologici del nostro
organismo i glicocorticoidi occupano un posto di rilievo in quanto si
dimostrano essenziali per controbilanciare gli effetti
pro-infiammatori di citochine, chemochine e altri, formati in
risposta a un attacco infettivo o di altra natura; la loro azione
regolatrice viene quindi ad essere analoga a quella di molti altri
sistemi, attivanti e inibenti, presenti negli umani: fattori
favorenti la coagulazione e fattori trombolitici, fattori adatti alla
digestione endocellulare delle proteine (tripsine) e fattori
antitripsina e così via.
Mentre l'infiammazione
localizzata è per lo più benefica, l'infiammazione
eccessiva o persistente può portare alla distruzione di
tessuto e alla malattia. Insieme ad altri disordini, come l'asma e
altre malattie allergiche, le malattie autoimmunitarie e le sepsi,
essa è una causa importante di malattia e di morte. I
glicocorticoidi sono indicati per la maggior parte di queste
affezioni; la loro efficacia nel curare i disordini infiammatori
deriva dall'effetto pleiotropico del recettore dei glicocorticoidi
su una molteplicità di vie metaboliche. Ma questo effetto
molteplice dei glicocorticoidi può determinare, oltre agli
effetti favorevoli, anche degli effetti contrari, come il ritardo di
crescita nei bambini, l'immunosoppressione, l'ipertensione,
l'inibizione della guarigione delle ferite, l'osteoporosi e le
alterazioni metaboliche. Tutte queste attività possono alla
lunga divenire pericolose per l'organismo, per cui esse
complessivamente controindicano l'uso prolungato dei
glicocorticoidi.
Di seguito verranno presi in considerazione i
meccanismi, con i quali i glicocorticoidi inibiscono l'infiammazione
e insieme le limitazione di questi ormoni in terapia.
Azioni basilari dei glicocorticoidi endogeni
L'asse
ipotalamo-ipofisi-surrene gioca un ruolo centrale nel regolare i
segnali sui recettori dei glicocorticoidi, presenti praticamente su
tutte le cellule. In breve, segnali nervosi, endocrini e citochimici
convergono a livello del nucleo periventricolare dell'ipotalamo per
controllare la secrezione dell'ormone liberante la corticotropina
(ACTH) nel sistema portale ipofisario. Ne consegue che la liberazione
dell'ormone liberante la corticotropina porta alla liberazione di
ACTH da parte dell'ipofisi anteriore. L'ACTH induce a sua volta
la sintesi e la secrezione di idrocortisone (cortisolo) da parte
della corteccia surrenale. La maggior parte del cortisolo secreto
(circa il 90%) è legato alle globuline, leganti i
corticosteroidi, nel sangue circolante. Il cortisolo libero è
la parte biologicamente attiva dell'ormone ed è convertito a
cortisone dalla 11-β-idrossisteroido-deidrogenasi, tipo 2. Al
contrario il tipo 1 di 11-β-idrossisteroido-deidrogenasi converte il
cortisone in idrocortisone.
Il recettore dei glicocorticoidi è
un membro della famiglia delle proteine che legano gli ormoni
steroidei. Esso si lega fortemente all'idrocortisone e promuove il
distacco dai recettori degli “chaperon” molecolari, incluse le
proteine heat-shock.
All'interno della cellula l'idrocortisone agisce in tre modi:
il complesso recettore-cortisolo muove verso il nucleo, dove si lega, come un omodimero (un complesso di due proteine identiche, legate da un ponte non covalente), alle sequenze del DNA, chiamate elementi glucocorticoidi-respondenti. Il risultante complesso agisce come attivatore o repressore di proteine che iniziano la trascrizione di alcuni geni da parte della RNA polimerasi II.
la stimolazione di altri geni riguarda l'interazione fra il complesso recettore-cortisolo e altri fattori di trascrizione, come il fattore-kB. Questa ultima azione avviene a bassi livelli di cortisolo.
il terzo meccanismo interessa i recettori, presenti sulla membrana cellulare e un secondo messaggero, chiamato “via non genomica”, perché agisce attraverso la stimolazione di parti, non corrispondenti a componenti nucleari.
I glicocorticoidi quindi inibiscono l'infiammazione attraverso questi 3 meccanismi, di cui due legati a effetti diretti e indiretti su alcuni geni, presenti nel nucleo della cellula, e uno dovuto a un effetto non legato ai geni (non genomico) (vedi figura 1 e figura 2).
Struttura del recettore dei glicocorticoidi
Il gene del recettore umano dei glicocorticoidi (GR) è un locus del cromosoma 5q31-32 (figura 3). Le variazioni nella struttura e nella espressione di questo gene portano a modificazioni nel segnale dei glicocorticoidi, per cui modificazioni nei diversi componenti di questo gene portano a dirette conseguenze sull'attività dei glicocorticoidi.
Regolazione neuroendocrina dell'infiammazione
L'interazione
fra sistema nervoso, asse ipotalamo-ipofiso-surrene e componenti del
sistema immune innato e adattivo, gioca un ruolo chiave nella
regolazione dell'infiammazione e della immunità. Per esempio
le citochine e i mediatori dell'infiammazione attivano i recettori
periferici del dolore, i cui assoni attraverso le corna dorsali del
midollo e le sinapsi con il tratto lemniscale, portano il segnale del
dolore al talamo e alla corteccia somato-sensitiva. L'attivazione
di questa via nocicettiva stimola infine l'attività
dell'asse ipotalamo-ipofisi-surrene.
I glicocorticoidi, che
vengono così sintetizzato, inibiscono a loro volta la sintesi
di citochine e di mediatori dell'infiammazione, venendo così
a formare un ansa feedback negativa. Le citochine possono agire anche
sul cervello, attivando direttamente l'asse
diencefalo-ipofisi-surrene. Una cattiva regolazione di questa ansa
neuroendocrina determina modificazioni generali nell'infiammazione
e nell'immunità.
L'iperattività dell'asse
ipotalamo-ipofisi-surrene in assenza d'infiammazione, come avviene
nella sindrome di Cushing, determina immunosoppressione e aumenta
quindi la suscettibilità alle infezioni. Il dolore fisico, il
trauma emotivo e la restrizione calorica attivano l'asse
ipotalamo-ipofisi-surrene e causano immunosoppressione. Anche la
diminuzione dell'attività dell'asse
ipotalamo-ipofisi-surrene e bassi livelli di glicocorticoidi
aumentano la suscettibilità alle infezioni e la gravità
stessa dell'infiammazione. I pazienti con malattia di Addison, per
esempio, necessitano della somministrazione di glicocorticoidi
durante le infezioni e l'infiammazione per prevenire l'effetto
dannoso delle citochine. La cattiva regolazione dell'asse
ipotalamo-ipofisi-surrene da parte dell'infiammazione si associa a
una cattiva prognosi in pazienti con sindrome da difficoltà
respiratoria acuta. Analogamente la resistenza acquisita ai
corticosteroidi è un evento comune nei pazienti con artrite
reumatoide cronica per una diminuita espressione del recettore α dei
glicocorticoidi, per un'aumentata espressione del recettore α o per
in attivazione della chinasi proteica attivata dai fitogeni (MAPK),
che fosforilizza il recettore dei glicocorticoidi e inibisce quindi
la capacità segnalante di questi ormoni.
Figura
1 – Vie di comunicazione fra il sistema nervoso, l'asse
ipotalamo-ipofisi-surrene e altri tessuti, influenzati dai segnali
dei corticosteroidi.
(fonte: Rhen T, Cidlowski JA. N.Engl J Med
2005, 353:1711-23) 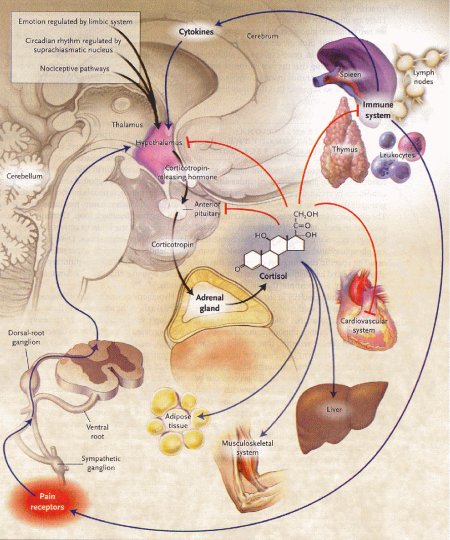
Le frecce rosse dimostrano inibizione e le frecce blu o nere indicano attivazione.
Meccanismi antiinfiammatori di segnalazione
I glicocorticoidi e i loro recettori sono situati all'apice di una rete regolatoria che blocca molte vie infiammatorie (vedi figura 4). Per esempio i glicocorticoidi possono inibire la produzione di prostaglandine attraverso tre meccanismi indipendenti:
induzione e attivazione dell'annessina 1
induzione della fosfatasi 1 MAPK
repressione della trascrizione della cicloossigenasi 2.
Figura
2 – I tre principali meccanismi di azione dei glicocorticoidi e dei
suoi recettori nell'inibizione dell'infiammazione.
(fonte:
Rhen T, Cidlowski JA. N.Engl J Med 2005, 353:1711-23)
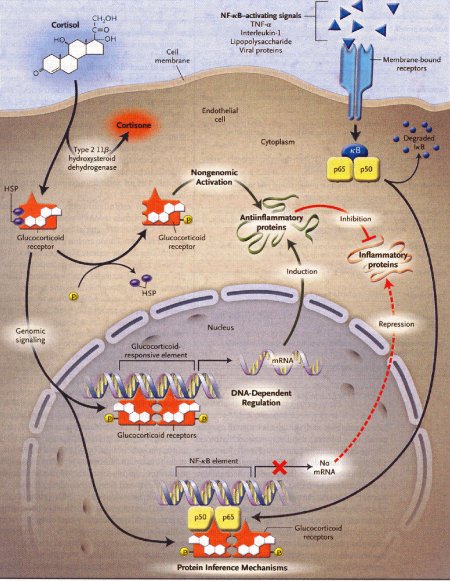
TNF-α =
tumor necrosis factor α
HSP = heat-shock protein
mRNA = RNA
messaggero
P = fosfati
Le frecce nere indicano attivazione, le
frecce rosse invece inibizione, il segno X indica mancanza di
produzione (blocco), cioè non produzione di mRNA.
Figura
3 – Localizzazione del gene e organizzazione del recettore umano
per i glicocorticoidi (GR).
(fonte: Rhen T, Cidlowski JA. N.Engl J
Med 2005, 353:1711-23)
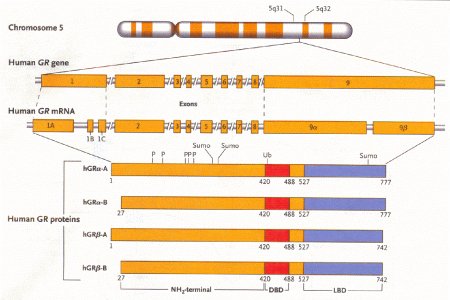
La
diversità nella espressione e nella funzione dei recettori dei
glicocorticoidi risulta dalla sede alternativa della
trascrizione.
DBP = indica il dominio legante il DNA
LBD =
indica il dominio legante il ligando
hGR = recettore umano dei
glicocorticoidi
L'
annessina 1 (chiamata anche lipocortina-1) è una proteina
anti-infiammatoria che fisicamente interagisce e inibisce una
fosfolipasi citosolica A2_ (cPLA2α). Questa
proteina legante il calcio richiede elevati livelli di calcio e
l'azione di fosforilazione da parte della proteinchinasi MAPK. II
calcio/calmodulina dipendente e la chinasi MAPK sono infatti
necessari perché essa eserciti la sua attività
enzimatica. L'attivazione della cPLA2 α da parte dello
stimolo infiammatorio inizia al momento della fosforilazione da parte
del citosol sulla membrana perinucleare, dove esso idrolizza i
fosfolipidi contenenti acido arachidonico. I glicocorticoidi inducono
e attivano l'annessina 1, che, inibendo la cPLA2α,
blocca la liberazione di acido arachinonico e la sua successiva
conversione in acido eicosanoide (cioè prostaglandine,
tromboxani, prostracicline e leucotrieni). I topi, mancanti di
annessina 1 hanno elevati livelli di cPLA2α , un'esagerata
risposta infiammatoria e una resistenza parziale all'azione
anti-infiammatoria dei glicocorticoidi. Esiste una forte correlazione
fra livelli di cortisolo basali, stimolati da parte dell'ACTH da un
lato e l'espressione dell'annessina 1 sui neutrofili umani
dall'altro; è sconosciuta l'importanza clinica
dell'annessina 1 come proteina anti-infiammatoria.
Una seconda
proteina anti-infiammatoria, indotta dai glicocorticoidi è la
fosfatasi 1 MAPK. Le citochine, le infezioni batteriche e virali e la
luce ultravioletta sono alcuni segnali infiammatori che attivano la
cascata MAPK. La fosfatasi MAPK defosforila e inattiva tutti i membri
della famiglia delle proteine MAPK, inclusa la chinasi Jun
N-terminale, le chinasi 1 e 2, dei segnali extracellulari, e la
chianasi p38. Di conseguenza la fosfatasi 1 MAPK può inibire
anche l'attività cPLA2α bloccando la
fosforilazione da parte della chinasi MAPK e MAPK interagente.
Inoltre i glicocorticoidi e il loro recettore interferiscono
direttamente con la trascrizione c-Jun mediata.
Figura
4 –Architettuta molecolare parziale riguardante l'antagonismo dei
glicocorticoidi con l'infiammazione
(fonte: Rhen T, Cidlowski
JA. N.Engl J Med 2005, 353:1711-23) 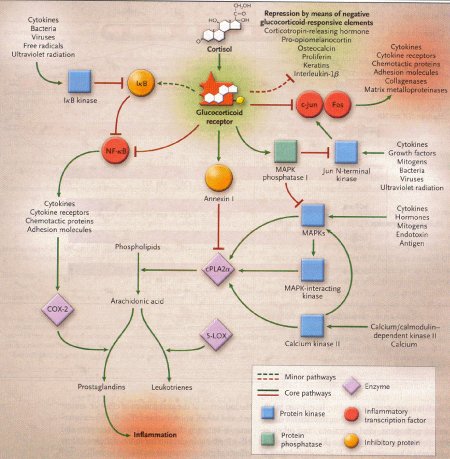
Le vie
infiammatorie sono caratterizzate da anse feedback positive.I
recettori dei glicocorticoidi inibiscono queste vie in diversi punti
sia bloccando la trascrizione delle proteine infiammatorie da parte
dell'NF-κB e proteina 1 attivatrice, sia inducendo l'espressione
di proteine anti-infiammatorie, come _B1, annessina 1 e fosfatasi 1
MAPK.
5-LOX = 5-lipodssigenasi; COX-2 = cicloossigenasi 2. Le
linee rosse significano inibizione e le frecce rosse attivazione.
Il
complesso del recettore del cortisolo interagisce anche fisicamente
con il NF-κB per bloccare la sua attività trascrizionale. Nel
suo stato inattivo NF-κB è sequestrato nel citoplasma da una
proteina inibitoria, chiamata I_B. Il tumor necrosis factor α,
l'interleuchina 1, gli agenti microbici, le infezioni virali e
altri stimoli infiammatori attivano la cascata che attiva le chinasi
I_B. La fosforilazione dell'I_B porta alla sua degradazione da
parte del proteasoma e alla liberazione dell'NF-κB. Nel nucleo,
l'NF-κB si lega alle sequenze del DNA, chiamate elementi NF-κB e
stimola la trascrizione delle citochine, delle chemochine, delle
molecole di adesione cellulare, dei fattori del complemento e dei
recettori di queste molecole. L'NF-κB induce inoltre la
trascrizione della ciclossigenasi 2, un enzima essenziale per la
produzione di prostaglandine. Così l'antagonismo del NF-κB,
indotto dai glicorticoidi, e la repressione della ciclossigenasi 2
rappresentano un ulteriore meccanismo d'inibizione della sintesi
delle prostaglandine, dopo l'induzione degli antagonisti del
cPLA2α, da parte dell'annessina 1 e della fosfatasi 1 MAPK (vedi
figura 4). L'interazione diretta fra recettore dei
glicocorticoidi e NF-κB probabilmente riguarda la maggior parte degli
effetti inibitori dei glicocorticoidi sull'azione segnalante del
NF-κB. I glicocorticoidi e i recettori dei glicocorticoidi modulano
anche l'attività di altri fattori di trascrizione.
Recenti
pubblicazioni suggeriscono che i glicocorticoidi possono avere un
rapido effetto sull'infiammazione, anche senza determinare
cambiamenti nell'espressione dei geni. Il meccanismo non genomico,
meglio descritto, interessa l'attivazione della sintetasi
dell'ossido nitrico endoteliale (eNOS). I glicocorticoidi stimolano
l'attività della fosfatidilinositol-3-idrossichinasi (PI3K)
in un recettore corticoido-dipendente, ma non
trascrizione-indipendente, presente sulle cellule endoteliali umane.
L'attivazione del PI3K porta alla fosforilazione dell'Akt. L'Akt
fosforilato fosforilizza e attiva eNOS, con la conseguente produzione
di ossido nitrico. Questo reperto è sorprendente perché
la produzione di ossido nitrico è generalmente associata a
vasodilatazione e infiammazione. Molte ricerche saranno necessarie
per chiarire il ruolo dei meccanismi non-transcrizionali
nell'inibizione della vasodilatazione, della permeabilità
vascolare e della migrazione dei leucociti attraverso
l'endotelio.
Risulta evidente da tutto questo che i
glicocorticoidi agiscono a diversi livelli mediante meccanismi
multipli per controllare l'infiammazione.
Limitazioni della terapia con glicocorticoidi
| Tessuto | Effetti collaterali |
|---|---|
| Surrene | Atrofia surrrenalica. Sindrome di Cushing |
| Sistema cardio-vascolare | Displipidemia, ipertensione, trombosi, vasculite |
| Sistema nervoso centrale | Modificazioni del comportamento, dell'apprendimento, della memoria e dell'umore (cioè psicosi), atrofia cerebrale |
| Apparato gastro-intestinale | Sanguinamento gastro-intestinale, pancreatine, ulcera peptica |
| Sistema immune | Ampia immunosoppressione, attivazione di virus latenti, |
| Cute | Atrofia, guarigione ritardata delle ferite, eritema, ipertrricosi, dermatite periorale, petecchie, acne indotta dai glicocorticoidi, strie rubre, teleangectasie |
| Sistema muscolo-scheletrico | Necrosi ossea, atrofia muscolare, osteoporosi, ritardo di crescita longitudinale delle ossa |
| Occhi | Cataratta, glaucoma |
| Rene | Aumentata ritenzione di sodio e aumentata escrezione di potassio |
| Sistema riproduttivo | Pubertà ritardata, ritardo di crescita fetale, ipogonsadismo |
Il trattamento prolungato con glicocorticoidi può causare ipertensione attraverso due meccanismi:
la ritenzione renale di sodio e quindi l'aumento del volume ematico
il potenziamento delle risposte vasopressorie all'angiotensina II e alle catecolamine.
L'aumentata risposta all'angiotensina II è dovuta all'induzione dei suoi recettori da parte dei glicocorticoidi. D'altra parte i glicocorticoidi non modificano il numero o l'affinità dei recettori _1-adrenergici, mentre invece potenziano i segnali _1-adrenergici.
Sebbene la resistenza vascolare sistemica, indotta dai glicocorticoidi, sia dannosa, modificazioni localizzate nella vasoreattività possono portare a effetti benefici con il trattamento combinato, glicocorticoidi + _2-agonisti, nei pazienti con asma. Un corso di due settimane di glicocorticoidi inalati infatti diminuisce la perfusione basale della mucosa polmonare e ristabilisce la risposta vascolare ai _2-agonisti in pazienti con asma.
La
crescita longitudinale nei bambini è il risultato della
proliferazione e differenziazione dei condrociti e la successiva
ossificazione della matrice extracellulare nelle placche di crescita
delle ossa lunghe. Le cellule staminali risiedono all'estremo
epifisario del piatto delle ossa lunghe e danno luogo alla
proliferazione dei condrociti. Quanto più si allontanano
dall'osso metafisario e quanto più i condrociti rallentano
la loro velocità di proliferazione, iniziano a ipertrofizzarsi
e producono le proteine della matrice extracellularre e le
metalloproteinasi della matrice. Mentre i condrociti preparano questa
impalcatura, essi assorbono calcio e secernono fosfato e
idrossiapatite. Infine essi vanno incontro ad apoptosi lasciando
dietro di loro l'osso mineralizzato. I glicocorticoidi rallentano
la crescita longitudinale riducendo la proliferazione dei condrociti
e determinando l'apoptosi di queste cellule. L'inibizione del
fattore 1 di crescita, insulino-simile, è uno dei meccanismi
attraverso i quali si manifesta la riduzione dei condrociti. Il
fattore 1 di crescita insulino-simile aumenta la fosforilazione
dell'AKT e agisce come un fattore di sopravvivenza dei condrociti
trattati con glicocorticoidi. Sebbene vi possa essere una ripresa
della crescita quando il trattamento corticosteroideo viene sospeso,
trattamenti prolungati durante l'infanzia e la fanciullezza spesso
si associano a diminuzione della statura dell'adulto.
I
glicocorticoidi hanno un effetto deleterio anche sull'osso
dell'adulto. Osteoporosi e aumentato rischio di fratture sono i
principali effetti collaterali del trattamento con glicocorticoidi.
L'osteoporosi in risposta ai glicocorticoidi è dovuta in
parte all'inibizione della trascrizione della osteocalcina negli
osteoblasti; l'osteocalcina è un'importante proteina della
matrice extracellulare, che promuove la mineralizzazione delle
ossa.
Riassumendo i glicocorticoidi aggravano l'osteoporosi
inducendo l'apoptosi degli osteoblasti e aumentando l'attività
degli osteoclasti.
I glicocorticoidi inibiscono anche la
guarigione delle lesioni asettiche. Per esempio le fratture inducono
l'infiammazione, mentre la produzione di citochine si ritiene sia
essenziale per la guarigione e il rimodellamento dell'osso. Oltre a
bloccare la sintesi delle citochine, i gloicocorticoidi inibiscono la
sintesi delle metalloproteinasi e del collageno della matrice,
fattori importanti per la guarigione delle ferite.
Inoltre i
glicocorticoidi promuovono la gliconeogenesi nel fegato, la
degradazione delle proteine in aminoacidi nel muscolo (atrofia) e la
lipolisi. Non ci sono al momento mezzi per migliorare gli effetti
collaterali della terapia prolungata con corticosterodi, sia a
livello dei recettori che degli elementi steroidi.-responsivi. Spesso
viene usato un trattamento con insulina per un diabete indotto dai
glicocosrticoidi o con bifosfonati per l'osteoporosi.
L'esistenza
di questi problemi ha indotto i ricercatori a identificare
glicocorticoidi potenzialmente selettivi.
Glicorticoidi selettivi e terapie del futuro.
E'
ormai chiaro che gli effetti anti-infiammatori dei glicocorticoidi
sono principalmente mediati dall'inibizione dell'NF-κB e della
proteina 1 attivatrice, mentre i loro effetti collaterali derivano
dall'attivazione della trascrizione.
Uno studio recente (Schacke
H et al, Proc Nat Acad Sci USA 2004, 101:227-32) ha descritto un
nuovo cortisonico (ZK216348) con un quadro di repressione e
attivazione della trascrizione, che è risultato
dranmaticamente differente da quello dei vecchi glicocorticoidi. Il
livello di glicocorticoide richiesto per reprimere l'interleuchina
8 nei monociti è da 8 a 12 volte superiore, quello richiesto
per indurre la tiroxina.-aminotranferasi nelle cellule
epatiche.
Esiste quindi la possibilità di sviluppare
selettivi glicocorticoidi dotati di un migliore profilo terapeutico.
Conclusioni
Gli
effetti anti-infiammatori e immunosoppressivi dei glicocorticoidi
utilizzano molti meccanismi molecolari, che sono stati chiariti
grazie alla ricerca di base.
Come abbiamo visto tre meccanismi
principali riguardano gli effetti dei corticosteroidi:
il legame dei cortocosterodi ai recettori, localizzati sugli elementi glicocorticoidi-responsivi (come l'induzione dell'annessina 1 e della MAPK fosfatasi 1),
gli effetti indiretti sull'espressione dei geni, attraverso l'interazione dei recettori dei glicocorticoidi su altri fattori di trascrizione (cioè NF-κB e attivatore della proteina 1)
e gli effetti (non-genomici) mediati dai recettori dei glicocorticoidi sulla cascata del secondo messaggero (cioè la via PI3K-Akt-eNOS).
Sfortunatamente poiché alcuni di questi meccanismi sono interessati anche ai segnali fisiologici, oltre che ai segnali anti-infiammatori, gli effetti terapeutici dei glicocorticoidi sull'infiammazione si accompagnano spesso a significativi effetti collaterali.
E' augurabile che si arrivi alla preparazione di glicocorticoidi provvisti di affetti benefici, senza presentare gli effetti sfavorevoli (Buttgereit et al. Lancet 2005, 365:801-3).