I difetti del tubo neurale
Clinica Pediatrica, IRCCS, Burlo Garofolo, Trieste
E mail:marzia_lazzerini@libero.it.
Le immagini sono tratte da un esperienza personale in Brasile presso l'istituto Materno Infantil de Pernambuco (IMIP).
La zona del Pernambuco, nel nord-est del Brasile, è tra le più povere del paese. L'accesso alle cure antenatali (prevenzione con acido folico, screening con misurazione dell'alfa-fetoproteina, ecografia fetale) è carente in particolare per motivi di ordine economico e sociale. Molte malformazioni rimangono misconosciute sino al momento del parto; i neonati vengono indirizzati subito dopo la nascita verso strutture di terzo livello senza che vengano instaurate le adeguate precauzioni per la prevenzione delle complicazioni (sovrainfezione). La legislazione brasiliana comunque non contempla l'interruzione volontaria di gravidanza per i feti con queste malformazioni.
Per questi motivi l'incidenza di bambini portatori di malformazioni gravi è particolarmente elevata, il che rafforza la necessità di conoscere a fondo l'evoluzione clinica e le possibilità terapeutiche per la gestione di questi casi.
I difetti del tubo neurale (Neural Tube Defects NTDs): Sono malformazioni risultati da difetti di sviluppo del tubo neurale, ovvero la struttura embrionaria che si forma per il ripiegamento e fusione di due estremità della piastra neurale a forma appunto di tubo, e che dà origine al sistema nervoso centrale (encefalo e midollo spinale). Comunemente si ritiene che i difetti del tubo neurale siano secondari a difetti di fusione (processo che inizia contemporaneamente in segmenti distinti e che si compie normalmente intorno al 29° giorno post-concepimento), piuttosto che alla riapertura di segmenti già precedentemente saldati.
Definizioni: esiste notevole variabilità nella nomenclatura, comunque generalmente si definisce quanto segue
Anencefalia: difetto di chiusura dell'estremità superiore del tubo neurale anteriore, con esposizione di tessuto encefalico emorragico e degenerato attraverso la perdita di continuità della callotta cranica. La mortalità è del 100%, 75% dei quali come aborti precoci.
Encefalocele: è in genere una lesione chiusa. Si definisce come erniazione del tessuto encefalico al di fuori della callotta cranica e deriva da difetti del mesoderma (in corrispondenza o subito dopo la chiusura del tubo neurale). Circa l'80% si verifica a livello occipitale.
Meningocele e Mielomeningocele: sono spesso anche definite comeSpina bifida cistica, nel senso di deficit di saldatura degli archi vertebrali posteriori con formazione di una tumefazione cistica esternamente visibile. Nell'80% dei casi si riscontrano a livello lombare e nell'80% dei casi sono rivestiti da cute non integra. I meningoceli (erniazione delle sole meningi Fig 1-2) generalmente non si associano a deficit neurologico, a differenza dei mielo-meningoceli (erniazione di meningi e midollo spinale Fig 3-6).
Spina bifida occulta: è una malformazione della parte caudale del midollo, con difetto di saldatura dell'arco vertebrale, ricoperto da cute integra. Si manifesta in forma benigna senza essere causa di patologia nel 10% della popolazione. Può associarsi alla presenza di una fossetta cutanea a livello sacrale, a volte con raccolta pilifera (seno dermico spinale Fig 7-8), rappresentante l'ultimo punto di separazione tra l'ectoderma di rivestimento ed il tubo neurale. A volte tuttavia alcune cellule di rivestimento rimangono incorporate nel tubo neurale e danno origine a tumori ectodermici (massa dermoide intramidollare). Altre volte alla spina bifida si associa una dilatazione cistica del canale midollare (mielocistocele).
Incidenza:
E' di 1: 1000 nati vivi (negli stadi embrionari l'incidenza è del 2.5%, la maggio parte esitano in aborti spontanei).
I difetti più frequenti sono la spina bifida occulta, l'anencefalia, il mielomeningolcele.
L'incidenza è lievemente maggiore per il sesso femminile (RR: 1.5), e per la razza Ispanica, per i figli di madri giovani (<20 anni, RR: 3-4) o particolarmente anziane e per le classi economiche disagiate.
Il rischio è maggiore nei figli di madri con diabete insulino dipendente (sino all'1%).
Il 95% dei bambini con NTDs nascono da coppie senza storia familiare di questi difetti. Il rischio in una coppia con parente di primo grado affetto è stimato dello 0.3-4%.
Cause riconosciute:
Nutrizionali: deficit A FOLICO, vitamina B12, ZINCO
Anomalie cromosomiche: trisomie 13 e 18, triploidie, traslocazioni bilanciate, e cromosomi ad anaello
Sindromi: i NTDs sono stati osservati come parti di alcune sindromi (descritte oltre 50 sindromi), alcune delle quali con eredità mendeliana. Un tipico esempio è la sindrome di Meckel-Gruber (autosomica recessiva), che si presenta encefalocele, microcefalia, polidattilia, rene policistico, ed altri difetti delle vie urinarie. In modelli animali sono state individuate alcune mutazioni specifiche (ex pax3) che sosterrebbero le teorie basate sulle influenze genetiche come fattori causali primari dei NTDs.
Teratogeni: Antifolici (A valproico, carbamazepina, fenitoina, fenobarbital, methotrexate), talidomide, nitrati. Iperglicemia in figli di madri diabetiche. Anche il piombo e gli eteri gli colici sono stati sospettati associati agli NTDs.
Screening Prenatale:
α-fetoproteina (AFP) materna: tra la 14-16 settimana di gestazione. Valori elevati (X2.5 i valori normali) hanno sensibilità del 90-100%, specificità del 96%, valore predittivo negativo del 99-100%, ma un basso valore predittivo positivo (legato alla bassa incidenza dei difetti).
Ecografia fetale: Estremamente sensibile e specifica in mani esperte; utile anche per valutare altri difetti associati.
α-fetoproteina (AFP) nel liquidi amniotico ed acetilcolinesterasi: sensibilità nei confronti della anenecefalia e spina bifida occulta del 100% se combinati assieme.
Gestione:
Anencefalia: terapia di supporto (mortalità 100% nelle prime 2 settimane).
Encefalocele:
1. Esame fisico per altre malformazioni (sindrome di Meckel-Gruber) ed esami strumentali (Eco, Tac, RMN).
2. Neurochirurgia: da valutare caso per caso in base dimensioni, sede, qualità del rivestimento cutaneo. Spesso è necessario uno shunt ventricolo-peritoneale per la compresenza di idrocefalo (50% dei casi).
3. Canceling e follow up a lungo termine: i deficit visivi sono molto comuni negli encefaloceli occipitali. Deficit motori ed intellettivi dipendono dalla sede e dalle dimensioni della lesione (>50%).
Mielomeningocele
1. Esame fisico per malformazioni associate e valutazione neurologica.
La valutazione neurologica è importante per determinare il livello della lesione midollare, da cui dipende in gran parte l'outcome motorio finale. La presenza dei riflessi sfinterici suggerisce l'integrità delle vie spinale ed è prognosticamente importante identificando i bambini a prognosi migliore .
Tabella 1. CORRELAZIONE TRA SEDE DELLA LESIONE, OBIETTIVITA' NEUROLOGICA E PROGNOSI SULLA DEAMBULAZIONE.
Livello della lesione | Innervazione | Sensibilità cutanea | Funzione sfintere | Riflessi | Potenziale di deambulazione | |
Toraco -Lombare | T12-L2 | Inguine= L1 Faccia antero superiora coscia= L2 | - | - | nullo | |
Lombare | L2-3 | Faccia antero inferiore coscia e ginocchio=L3 Faccia mediale gamba=L4 | - | Ginocchio | Possibilità deambulare con stampelle | |
Lombo -Sacrale | L5-S1 | Faccia laterale gamba e mediale piede=L5 Palma del piede=S1 | - | Caviglia | Possibilità deambulare con o senza stampelle | |
Sacrale | S2-4 | Faccia posteriore gamba ed inguine=S2 Natiche=S3,S4 | Riflessi vescicali e rettali funzionanti | anale | Possibilità di deambulare senza stampelle 2. Cure primarie: Copertura sterile + antibiotici 3. Valutazione strumentale 4. Correzione chirurgica: generalmente entro 24-48 h per scongiurare infezioni 5. Valutazione complicazioni:
Spina bifida occulta Le rare forme di spina bifida occulta associate a malformazioni maggiori si rendono evidenti progressivamente se non diagnosticate nel periodo neonatale con:
La diagnosi si avvale dell'imaging (RMN). Nelle forme acute il trattamento chirurgico tempestivo (entro 48h) con decompressione del midollo spinale può recuperare completamente o parzialmente la funzionalità. Tecniche di chirurgia intrauterina Per il mielomeningocele sono attualmente proposte tecniche di chirurgia intrauterina. Al momento attuale i criteri internazionalmente accettati per la definizine di candidato sono: a) mielomeningocele isolato; b) assenza di anomalie cromosomiche; c) età gestazionale tra le 20 e 25 settimane. Bibliografia- Gomella TL et al- Neonatology. 4° Ed. Lange Medical Books/Mc Graw Hill 1999. - Botto LD, Moore CA, Khoury MJ, Erickson JD. Neural-tube defects. N Engl J Med 1999;341(20):1509-19. - Frey L, Hauser WA. Epidemiology of neural tube defects. Epilepsia 2003;44 Suppl 3:4-13 - Sbragia L, Machado IN, Braidos Rojas CE, Zambelli H, Lopes Miranda M, Bianchi MO, Barini R.. Fetal myelomeningocele and the potential in-utero repair: follow-up of 58 fetuses. Arq. Neuro-Psiquiatr. 62 (2) 2004 - Mitchell LE, Adzick NS, Melchionne J, Pasquariello PS, Sutton LN, Whitehead AS. Spina bifida. Lancet. 2004;364(9448):1885-95 | Figure 1-2
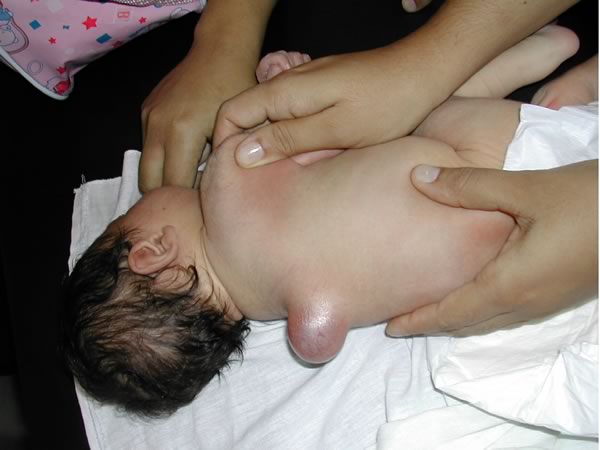
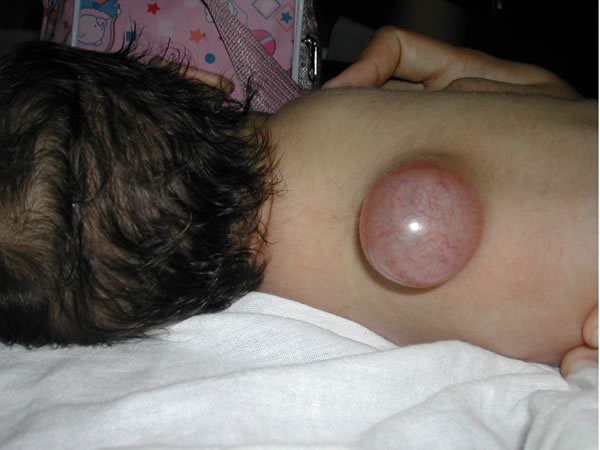
Meningocele a livello toracico con cute integra. Il bambino si presentava senza deficit neurologici Figure 3-4
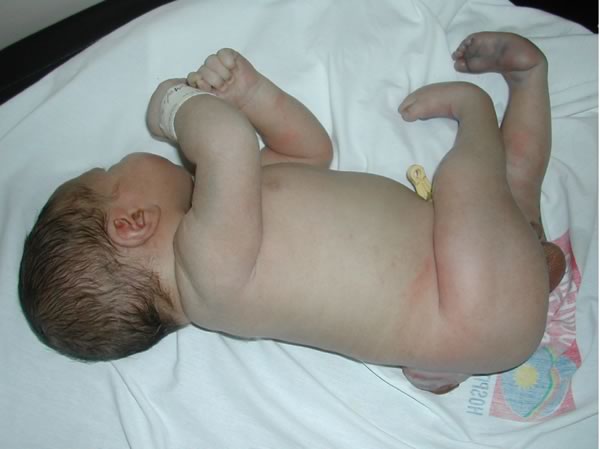
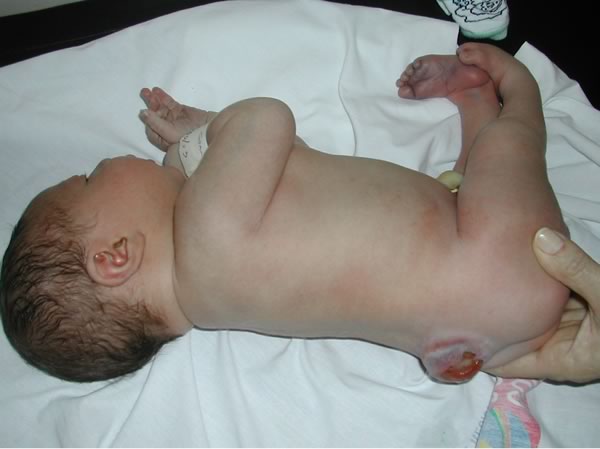
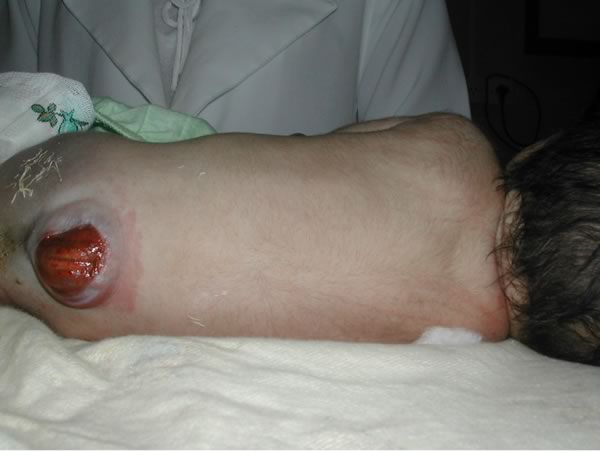
Mielo-meningocele a livello lombo-sacrale con cute lesa. E' evidente il deficit neurologico a livello degli arti inferiori, con mioatrofia dei segmenti distali e piede torto bilaterale. Figure 5-6
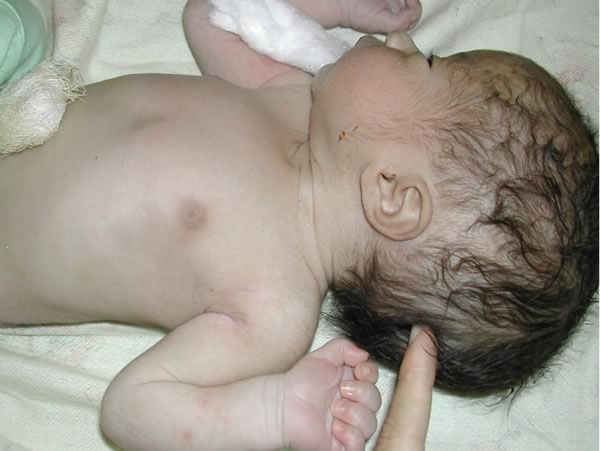
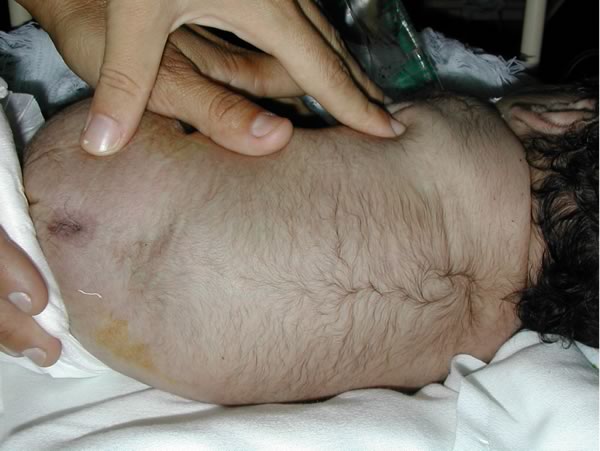
Mielomeningocele ed idrocefalo. Bambino con esposizione di meningi e midollo in sede sacrale. Era inoltre presente macrocefalia e diastasi delle suture, in un quadro di idrocefalo. Figure 7-8
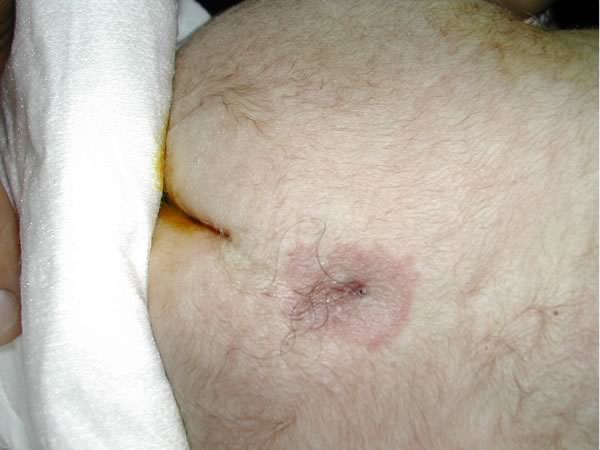
Sospetta spina bifida occulta. Bambina polimalformata (macrosomia, facies particolare, cardiopatia, piede torto) con seno dermico spinale |