Cifoscoliosi, ipotonia e iperlassità ligamentosa alla nascita: a quale sindrome pensare?
Scuola di Specializzazione in Pediatria, Università di Trieste
Indirizzo per corrispondenza: lorenza.matarazzo@gmail.com
La piccola A., secondogenita di genitori consanguinei di origine macedone, ci colpisce alla nascita per una severa cifoscoliosi. A un esame obiettivo più accurato emergono anche un’ipotonia generalizzata, un’iperlassità articolare, un’iperelasticità cutanea e l’assenza dei frenuli linguale e labiale inferiore. La gravidanza era decorsa regolarmente e non era stata segnalata alcuna anomalia perinatale. La madre ci racconta, solo dopo nostre precise domande, che anche alcuni cugini del padre sono affetti da grave cifoscoliosi. Valorizzando l’ipotonia e l’iperlassità ligamentosa, ipotizziamo possa trattarsi della forma cifoscoliotica della sindrome di Ehlers-Danlos(EDS); la diagnosi viene successivamente confermata dall’analisi genetica che evidenzia una duplicazione degli esoni 10-16 a carico del gene PLOD1, in omozigosi.
La EDS comprende un gruppo eterogeneo di malattie ereditarie del tessuto connettivo, caratterizzate da iperlassità articolare, cute iperelastica e fragilità tissutale.
Il tipo cifoscoliotico (EDS VI), estremamente raro (incidenza 1:100.000), è caratterizzato da scoliosi congenita progressiva, ipotonia muscolare grave, iperlassità articolare generalizzata e fragilità dei globi oculari. L’assenza dei frenuli linguale (sensibilità del 71,4%; specificità del 100%) e labiale inferiore (sensibilità del 100% e specificità del 99,4%) si associa alla EDS e può essere utile per identificare precocemente i neonati affetti.
La EDS VI è dovuta al deficit dell'enzima lisil-idrossilasi, codificato dal gene PLOD1, coinvolto nelle modifiche post-traduzionali della lisina, nelle procatene alfa del collagene, con conseguente instabilità dei tessuti affetti.
La diagnosi può essere effettuata attraverso il dosaggio delle piridinoline urinarie e confermata dall’analisi genetica (descritte 33 mutazioni diverse del gene PLOD1).
L'analisi molecolare del gene PLOD1 sui villi coriali può essere utilizzata per la diagnosi prenatale.
La presa in carico è esclusivamente sintomatica. La prognosi è variabile, e può essere grave, con perdita della deambulazione durante la seconda o terza decade di vita.
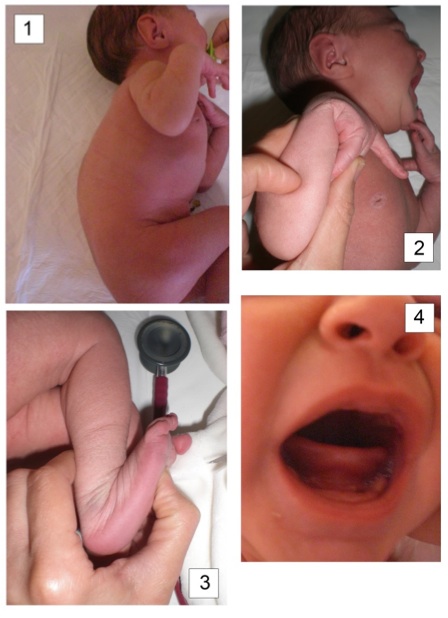
Figure. 1. Cifoscoliosi severa; 2. e 3. Iperlassità articolare; 4. Assenza del frenulo linguale.