Pigmenti fuori posto: indizi per una diagnosi rara
1Scuola di Specializzazione in Pediatria, Università di Bologna
2UOC Neuropsichiatria dell’Età Pediatrica, IRCCS “Istituto delle Scienze Neurologiche di Bologna”, Policlinico di Sant’Orsola
Indirizzo per corrispondenza: ezio.sarno@studio.unibo.it
Caso clinico
Una bambina di 1 anno accede in Pronto Soccorso Pediatrico (PSP) per episodio febbrile associato a esantema del tronco e in regione genitale, caratterizzato da elementi iperemici, pruriginosi e infiltrati, associati a elementi ipercromici. Dopo valutazione dermatologica tale dato viene inizialmente inquadrato come mastocitosi cutanea. I successivi controlli hanno documentato un buon controllo delle lesioni e gli elementi ipercromici sono stati inquadrati come un esito post-infiammatorio del quadro clinico iniziale.
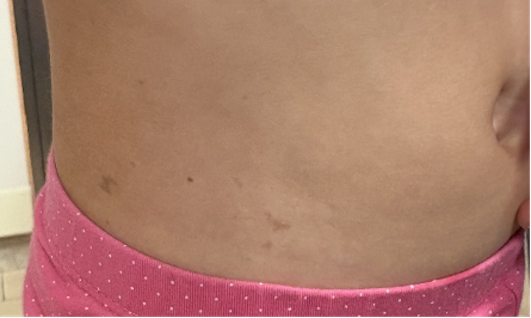
A distanza di tre anni accede nuovamente in Ambulatorio di Dermatologia pediatrica per la persistenza delle suddette lesioni, lievemente iperpigmentate a livello del tronco, dei fianchi (Figura), delle grandi labbra e della regione anteriore degli arti inferiori.
Durante l’esame obiettivo, inoltre, si riscontra piccola chiazza di alopecia cicatriziale della regione parieto-occipitale.
La piccola presenta anche agenesia di 3 elementi dentali e ritardo di acquisizione del linguaggio, per cui è già in carico presso il servizio di NPI territoriale e in attesa di intraprendere un percorso logopedico. In anamnesi la madre riferisce che anche lei, in età infantile, presentava un quadro dermatologico analogo a quello della figlia, andato incontro a risoluzione spontanea con persistenza di minimi esiti a livello cutaneo, contestuale all’agenesia di alcuni denti. Alla luce del quadro clinico e anamnestico, nel sospetto di incontinentia pigmenti, viene eseguita valutazione neuopsichiatrica infantile che ha confermato il sospetto clinico, e ha posto indicazione a un ricovero programmato per eseguire accertamenti a scopo di completamento diagnostico, comprensivi di valutazione genetica.
Durante il ricovero viene eseguita una RM encefalo con riscontro di focolai di gliosi nella sostanza bianca profonda, sia in sede periventricolare che a livello della corona radiata. Effettuate inoltre valutazione oculistica e polisonnografia, entrambi nella norma. Le indagini genetiche, eseguite sia sulla piccola che sulla madre, hanno evidenziato una delezione in eterozigosi (a carico di un solo cromosoma X) nelle regioni genomiche esoni 4-10 del gene IKBKG.
Tale delezione risulta causativa del quadro clinico descritto e l’esito degli esami è da considerare patognomonico e ha permesso di confermare a livello molecolare la diagnosi di incontinentia pigmenti. La bambina ha proseguito il suo follow-up NPI e dermatologico e a completamento diagnostico ha anche effettuato una valutazione cardiologica, risultata nella norma.
Discussione
Il caso clinico descritto rappresenta un caso esemplare di incontinentia pigmenti (IP), una rara displasia neuroectodermica a ereditarietà X-linked dominante, causata da mutazioni nel gene IKBKG/NEMO1. Tale gene codifica per una proteina essenziale nella regolazione del fattore nucleare NF-κB coinvolto in numerose funzioni cellulari, tra cui risposta immunitaria, sviluppo ectodermico, adesione cellulare e protezione contro l’apoptosi indotta da TNF. La mutazione causa ridotta attività di NF-κB e aumenta la suscettibilità all’apoptosi, con conseguenze severe nei maschi (spesso letali già in epoca fetale) e manifestazioni cliniche variabili nelle femmine. Dal punto di vista clinico, l’IP è una patologia multisistemica, con il coinvolgimento principale della cute, ma potenzialmente anche di SNC, occhi, denti, capelli, unghie, scheletro e apparato cardiopolmonare. La presentazione cutanea rappresenta il criterio diagnostico principale: le lesioni compaiono tipicamente nel primo anno di vita e si sviluppano lungo le linee di Blaschko. Sono classificate in quattro stadi:
- vescicolo/bolloso
- verrucoso/ipercheratosico
- iperpigmentato (fase tipica della IP) e
- atrofico/ipopigmentato.
Non tutti i pazienti attraversano tutti e quattro gli stadi e spesso più stadi coesistono. La presenza di alopecia cicatriziale e agenesia dentale, come osservato nel caso riportato, rappresenta un coinvolgimento ectodermico tipico della malattia. Anche il ritardo dello sviluppo del linguaggio e le alterazioni documentate alla RM encefalo sono tipici del potenziale coinvolgimento neurologico, che può variare da esiti lievi a gravi disabilità cognitive o motorie. Il coinvolgimento oculare è riportato tra il 50% e il 77% dei casi: le alterazioni più gravi riguardano la retina (distacco retinico, aree avascolari) e possono condurre a cecità. Anche il coinvolgimento cardiopolmonare è documentato, sebbene meno frequente, e comprende difetti strutturali o fibrosi endomiocardica. In singoli casi, sono descritte anche alterazioni scheletriche.
La diagnosi differenziale dipende dallo stadio clinico. Nella fase vescicolo/bollosa, può includere infezioni neonatali (herpes simplex congenito, varicella), epidermolisi bollosa o pemfigoide bolloso. Nelle fasi iperpigmentate o atrofiche, è necessaria la distinzione da nevus epidermico lineare, iperpigmentazioni neviformi lineari o a vortice, ipomelanosi tipo Ito o vitiligine, che generalmente non presentano la fase infiammatoria iniziale né interessamento degli annessi cutanei.
L’approccio diagnostico attuale si basa su una combinazione di valutazione clinica, storia familiare e conferma molecolare mediante analisi del gene IKBKG/NEMO. La conferma della mutazione permette di stabilire con certezza la diagnosi, di identificare portatori familiari e di pianificare la consulenza genetica, fondamentale per la gestione della malattia e per la prevenzione dei rischi riproduttivi, considerando la letalità nei maschi e la probabilità del 50% di trasmissione alle figlie femmine.
Il trattamento è prevalentemente di supporto e multidisciplinare. Le lesioni cutanee possono risolversi spontaneamente ma, in fase acuta, possono essere utilizzati corticosteroidi topici o tacrolimus topico per rallentarne la progressione. La gestione delle complicanze neurologiche, oculari o scheletriche richiede valutazioni specialistiche periodiche e interventi mirati (ad esempio riabilitazione logopedica per i disturbi dei linguaggi, terapia oftalmologica per la retinopatia). Il follow-up deve essere individualizzato, con monitoraggi più frequenti nei primi anni di vita e successivamente periodici a seconda degli apparati coinvolti.
Conclusioni
Il caso descritto evidenzia come un’attenta osservazione clinica, il riconoscimento dei segni cutanei caratteristici, la valutazione neuropsichiatrica e l’approfondimento genetico siano fondamentali per una diagnosi precoce dell’IP e per la gestione multidisciplinare di questa malattia multisistemica rara.
La storia familiare rappresenta un elemento chiave per sospettare la malattia, soprattutto in presenza di lesioni cutanee e anomalie dentarie ereditarie.
La conferma molecolare mediante analisi genetica permette una diagnosi definitiva e guida il follow-up multidisciplinare, indispensabile per ridurre il rischio di complicanze neurologiche, oculari e dentarie.
Il trattamento precoce del ritardo di sviluppo e il monitoraggio periodico dei principali organi bersaglio sono fondamentali per garantire una buona qualità di vita nei pazienti affetti da IP.
Bibliografia
- Cammarata-Scalisi F, Fusco F, Ursini MV. Incontinentia Pigmenti. Actas Dermo-Sifiliográficas (English Edition) 2019;110(4):273-8. DOI: 10.1016/j.adengl.2019.03.009.