Non è mai troppo tardi...per fare una diagnosi
Divisione di Pediatria 2, Dipartimento di Medicina della Procreazione dell’Università di Pisa, Azienda Ospedaliera-Universitaria Pisana, Pisa ed Epatologia e Centro Trapianto di Fegato Pediatrico, UPMC-Ismett, Palermo
Questa è la storia di una diagnosi tardiva ma comunque cercata con ostinazione. Lorenzo è un bambino di 15 mesi, che negli ultimi due ha presentato ricorrenti episodi di vomito associati ad astenia e sonnolenza per cui sono stati effettuati alcuni esami ematochimici con il riscontro di una elevazione delle aminotrasferasi.
Lorenzo è unigenito di genitori non consanguinei, nato a termine da gravidanza fisiologica ed espletata con parto eutocico. Il bambino ha goduto di buona salute fatta eccezione per un rallentamento fino a un vero e proprio arresto della crescita a partire dai 12 mesi di vita. All’esame obiettivo Lorenzo appare in mediocri condizioni generali, vigile e reattivo, apiretico, anitterico con un’epatomegalia omogenea di consistenza aumentata. Gli esami effettuati confermano l’elevazione delle aminotransferasi (AST 11 x N; ALT 15 x N) che si associa a una marcata alterazione del profilo coagulativo (attività di protrombina 30% INR 2.3, fattore V 48%) e a una ammoniemia normale. Per il rapido peggioramento dell’emostasi, Lorenzo viene trasferito d’urgenza presso un centro di trapianto epatico pediatrico (Ismett di Palermo) in vista di un possibile trapianto di fegato in urgenza.
All’ingresso in Ismett, Lorenzo presenta un quadro di insufficienza epatica a esordio subacuto (attività di protrombina 17%), in assenza di ittero (bilirubina tot. 0,41 mg/dl) con altri parametri sintetici discretamente conservati (albumina 3,4 g/dl, colesterolo tot. 102 mg/dl, pseudocolinesterasi 9200b U/l). Nel dubbio di una possibile intolleranza ereditaria al fruttosio (HFI) viene messo a dieta priva di fruttosio; viene inoltre immediatamente avviato il programma di accertamenti necessari per inserire il piccolo in lista d’attesa per il trapianto. Viene inoltre posizionato un catetere venoso centrale mediante cui è regolarmente somministrato plasma fresco congelato.
Per quanto concerne l’indagine eziologica, sono escluse le principali cause infettive (HAV, HBV, HCV, EBV, CMV, HSV 1-2, VZV, Toxoplasma, Leishmania; negativa la ricerca del DNA per, Adenovirus e Parvovirus B19), tossiche (paracetamolo) e immunologiche (normale profilo delle immunoglobuline, test di Coombs diretto negativo, negativa la ricerca di ANA, SMA, LKM, ANCA, SLA, LC-1). L’attenzione viene quindi rivolta a possibili cause metaboliche: l’analisi genetica del gene aldolasi B per le 7 mutazioni più comuni risulta negativa escludendo quindi l’ipotesi di una HFI; i risultati dell’analisi del profilo delle acilcarnitine, degli amminoacidi plasmatici, degli acidi grassi a lunga catena e della Carnitina libera sono nella norma.
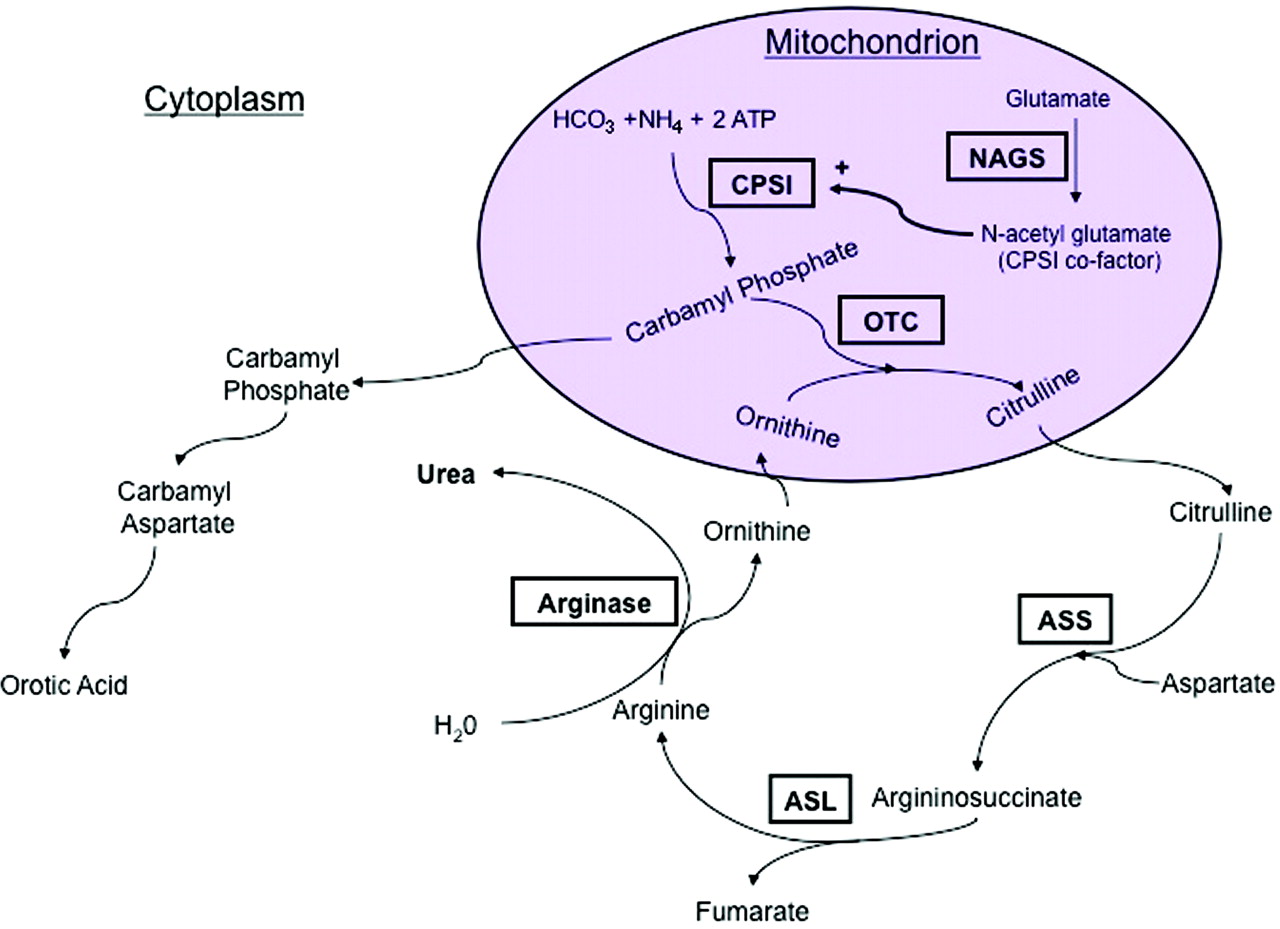
Figura. ASL = argininosuccinic acid lyase; ASS = argininosuccinic acid synthetase; CPSI = carbamoyl phosphate synthetase I; NAGS = N-acetylglutamate synthetase; OTC = ornithine transcarbamylase.
Sulla base del riscontro di una elevazione dell’acido orotico urinario (64 mmol/mol creatinina) in un campione di urine, durante il breve ricovero a Pisa (suggestiva di un deficit del ciclo dell’urea) viene intrapresa un’alimentazione ipoproteica/ipercalorica, affiancata ad una nutrizione parenterale notturna, al fine di raggiungere l’apporto calorico desiderato (120-130 Kcal/kg con 6 g/die di proteine). In più occasioni però, la ricerca di una conferma dei valori di una elevata aciduria orotica è negativa con una ammoniemia costantemente normale e comunque mai > 72 mmol/l. Tuttavia, nei giorni immediatamente successivi all’inizio della dieta ipoproteica, l’attività protrombinica migliora rapidamente tanto da permettere, l’esecuzione di una biopsia epatica percutanea che mostrerà un parenchima epatico con architettura lobulare conservata, pressoché privo di infiltrato infiammatorio, con epatociti lievemente polimorfi, ampio citoplasma chiaro e minimo accumulo di glicogeno. Un reperto istopatologico, in sostanza, compatibile con alterazioni epatiche da malattia metabolica. Il proseguimento della dieta ipoproteica/ipercalorica associata a supplementazione con Arginina per bocca, determinerà una totale e duratura normalizzazione del profilo emocoagulativo. Lorenzo viene quindi dimesso, senza diagnosi certa, per proseguire a domicilio un regime dietetico a bilancio proteico controllato.
Dall’agosto 2008 Lorenzo sarà seguito ambulatorialmente a Pisa con una progressiva liberalizzazione della dieta ipoproteica (molto mal tollerata) che sarà completata solo nel settembre 2010 (dopo circa 24 mesi). Lorenzo presenta comunque in questi due anni una crescita staturale intorno al 10° centile e un peso intorno al 3° centile per l’età, una persistente normalità del bilancio epatico, dell’emostasi, dell’ammoniemia e dell’acido orotico urinario. La ricerca delle mutazioni del gene della Ornitina Trans Carbamilasi (OTC) sarà negativa.
Nessuna anomalia clinica e di laboratorio sarà poi riscontrata fino a oltre 12 mesi dalla completa liberalizzazione della dieta, quando, a un controllo routinario, viene nuovamente evidenziata e confermata, una anomalia dell’emostasi (attività di protrombina 44%; aPTT 45 sec Fibrinogeno 172 mg/dl INR 1.68) in assenza di significate alterazioni del quadro clinico, della ammoniemia e degli enzimi epatici, ma questa volta nuovamente in presenza di una aciduria orotica patologica. Lorenzo verrà rimesso prudenzialmente a un regime ipoproteico (2 gm/kg) con una rapida normalizzazione dell’emostasi. Questa volta una nuova ricerca delle mutazioni del gene OTC identificherà una mutazione patogenetica.
Il deficit di ornitina transcarbamilasi è il più comune dei difetti del ciclo dell’urea con un’incidenza di 1:14.000 e una trasmissione X-linked. La presentazione clinica nei maschi avviene generalmente nel periodo neonatale con iperammoniemia acuta e coma che può risultare letale se non tempestivamente trattata. Nelle femmine eterozigoti e nei maschi in cui il difetto genetico determina solo una riduzione dell’attività enzimatica, il deficit di OCT può presentarsi in epoche della vita diverse e con una grande variabilità clinica tanto che è stimato che ben il 60% dei deficit di OTC non vengono poste in età non neonatale e nell’infanzia, ma anche in età adulta.
Bibliografia di riferimento
Smith W, Kishnani PS, Lee B, et al. Urea Cycle Disorders: Clinical Presentation Outside the Newborn Period. Crit Care Clin 2005;21:S9-S17.