Neurofibromatosi tipo I (parte prima)
Membro della Commissione Nazionale Vaccini
Indirizzo per corrispondenza: bartolozzi@unifi.it
La neurofibromatosi (una volta chiamata malattia di Recklinghausen), è un'affezione autosomica dominante alla quale il pediatra pensa con una certa frequenza, anche se la sua incidenza è solo di un caso su 3000 (un pediatra, con 80 nuovi nati ogni anno e con una carriera di 40 anni, in media può averne visto un solo caso).
Sono le macchie caffè e latte che richiamano alla mente del curante, la possibilità di una neurofibromatosi, anche se esse non sono così numerose come dovrebbero e anche se non hanno i caratteri classici di riconoscimento. Purtroppo capita a volte che, sull'onda della soddisfazione per una diagnosi difficile, il pediatra comunichi ai genitori i suoi sospetti diagnostici e quindi inserisca nella loro mente il dubbio che il proprio figlio sia affetto da una malattia grave, purtroppo non curabile. Poiché non esistono esami che siano in grado di escludere o di confermare al 100% la diagnosi, il dubbio nella mente dei genitori rimane. Si arriva così, spesso senza confermare o escludere la malattia, alle visite specialistiche del dermatologo, dell'oculista, del neurologo pediatra, del genetista e così via.
Per questo ho letto con interesse una Rivista comparsa sul Pediatrics di Gennaio 2009, di cui penso di riportare i punti salienti per impostare bene e risolvere il quesito diagnostico prima di parlarne apertamente con i genitori (Williams VC, Lucas J, Babcock MA, et al. Neurofibromatosis type I revisited. Pediatrics 2009;123:124-33).
La neurofibromatosi tipo I (NF1) è una delle più comuni malattie autosomico-dominanti del sistema nervoso, con un'incidenza di un caso su 2500-3000 soggetti, indipendentemente dalla etnia e dal sesso. La malattia è proteiforme: nella maggioranza dei casi tutto si limita alle macchie caffè e latte (75% dei casi), ma nel 25% dei pazienti si sviluppano tumori che vanno dai neurofibromi cutanei ai tumori del sistema nervoso, ai feocromocitomi fino ai fibrosarcomi. La malattia è dovuta a un'alterazione della differenziazione delle cellule della cresta neurale durante gli stadi precoci dell'embriogenesi, probabilmente come conseguenza dell'alterazione del fattore di crescita nervosa o del fattore di crescita gliale.
Usualmente la diagnosi si basa almeno due dei seguenti sette aspetti clinici, stabiliti nel 1987 dal National Institutes of Health Consensus Development Conference:
Macchie caffè e latte (> 0,5 cm al diametro maggiore nei prepuberi e > 1,5 cm in soggetti dopo la pubertà);
Numerose aree iperpigmentate di 2-3 mm di diametro (efelidi) alle pieghe cutanee (ascella e inguine);
Noduli di Lisch (amartomi dell'iride);
Due o più neurofibromi o > 1 neurofibroma plessiforme;
Gliomi delle vie ottiche;
Lesioni ossee caratteristiche (displasia dello sfenoide, curvatura della tibia e della fibula, pseudoartrosi degli arti inferiori, cifosi, scoliosi);
Familiarità di primo grado con neurofibromatosi tipo I (NF1), diagnosticata secondo questi criteri. Da ricordare che nel 30-50% dei casi si tratta di nuove mutazioni.
Aspetti clinici
| Aspetti clinici | Valutazione | Trattamento |
|---|---|---|
| Macchie caffè e latte | Aspetto diagnostico precoce (0-2 anni); macule iperpigmentate, della grandezza di 1-3 cm di diametro | Nessuna prova che possano essere rimosse con il laser; possono essere attenuate con il mascheramento |
| Efelidi | Aspetto diagnostico a 3-5 anni di età; frequenti alle ascelle e all'inguine con tendenza a diffondere | Non è necessario seguirne il decorso |
| Noduli di Lisch | Amartomi pigmentati dell'iride all'esame con la lampada a fessura, evidenti a 5-10 anni di età | Mandare all'oculista pediatra |
| Neurofibromi cutanei | Aspetto diagnostico del bambino e dell'adolescente: tumori benigni della guaina dei nervi periferici; possono presentare prurito localizzato | Rimozione chirurgica delle lesioni che danno dolore o che sfigurano il paziente, dipendendo dalla sede e dalla localizzazione; il laser CO2 può aiutare a rimuovere le lesioni superficiali più piccole; si possono avere delle ricadute, delle cicatrici ipertrofiche e dei deficit neurologici |
| Neurofibroma plessiforme | Aspetto diagnostico: tumori della guaina dei nervi periferici, che interessano molti fasci di nervi; possono estendersi alle strutture vicine con varie velocità di crescita e di aspetto: lesioni profonde possono essere evidenziate solo all'esame radiologico | Follow-up regolare con speciale attenzione ai sintomi e ai segni di trasformazione in tumori maligni delle guaine dei nervi periferici; la tomografia a emissione di positroni con fluorodesossiglucosio può distinguere i tumori benigni dai maligni; asportazione chirurgica per i tumori sintomatici; è controindicata la radioterapia |
| Tumori maligni della guaina dei nervi periferici | Spesso insorgono dai neurofibromi plessiformi; consultare uno specialista di NF1 se c'è un dolore persistente o un dolore che disturba il sonno o per la comparsa di nuovi e inspiegabili deficit neurologici (variazioni del tessuto da molle a duro) o aumento rapido della grandezza del neurofibroma | L'ideale è la resezione completa del tumore quando il tumore sia localizzato; la radioterapia è utile se la resezione è parziale, aggressiva o se il tumore è > 5 cm; i benefici della chemioterapia come secondo aiuto è controverso; follow-up ogni 3-4 mesi per le metastasi (tomografia computerizzata del torace, dolore osseo) |
| Displasia scheletrica | Vanno ricercati la scoliosi, i difetti ossei congeniti che facilitano la pseudoartrosi, la displasia dell'ala dello sfenoide, la macrocefalia, la bassa statura; seguire strettamente le curve di crescita e i parametri scheletrici | La scoliosi può richiedere il busto, la chirurgia correttiva con fusione vertebrale, a seconda della gravità; le pseudoartrosi rispondono poco alla chirurgia e può essere necessaria l'amputazione, sebbene una terapia precoce con bifosfonati possa essere benefica |
| Glioma delle vie ottiche | Risonanza magnetica o segni e sintomi endocrinologici | Risonanza magnetica a 3-12 mesi d'intervallo con valutazione neuro-oftalmica ed endocrinologica; chemioterapia standard con carboplatino e vincristina; la radioterapia è controindicata |
| Deficit neurocognitivi | Valutazione dello sviluppo neuropsicologico prima dell'inizio della scuola | Sviluppare un piano di educazione individuale; ottenere ogni anno una valutazione e un follow-up con uno speciale coordinatore delle necessità educative |
| Alterazioni cardio-vascolari | Valutazione annuale della pressione arteriosa e del cuore | Arteriografia renale ed escrezione delle catecolamine e dei metaboliti totali urinari nelle 24 ore per la pressione arteriosa: inviare i pazienti con soffi al cardiologo pediatra. |
Alterazioni |
Valutazione annuale della pressione arteriosa e del cuore
Arteriografia renale ed escrezione delle catecolamine e dei metaboliti totali urinari nelle 24 ore per la pressione arteriosa: inviare i pazienti con soffi al cardiologo pediatra.
Macchie caffè e latte
Sebbene esse possano essere presenti alla nascita, usualmente si sviluppano dai primi mesi di vita ai due anni di età. La loro comparsa è spesso il primo aspetto delle NF1 (Figura 1). Il numero delle macchie può essere elevato, fino a molte dozzine, ma né il loro numero né la loro grandezza sono legati alla gravità della malattia. La grandezza e il numero di queste macchie è invece importante per la diagnosi: la presenza di ≥ 6 macchie caffè e latte di > 0,5 cm di diametro prima della pubertà o di 1,5 cm di diametro dopo la pubertà è il primo criterio diagnostico (Figura 2). Le macchie caffè e latte non hanno tendenza a una trasformazione maligna. Per i pazienti che sono disturbati dalla loro presenza, è possibile dare dei consigli su come camuffarle. D'altra parte non ci sono prove a favore delle terapia laser per la loro rimozione.
Figura 1. Manifestazioni cliniche della NF 1 in merito alla loro età di comparsa
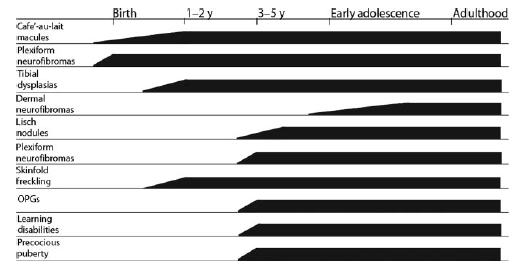
OPG = glioma del nervo ottico
Figura 2. Aspetti clinici della neurofibromatosi.
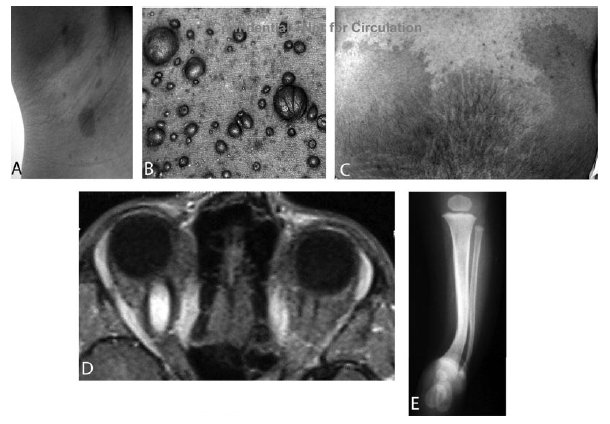
Le efelidi alle ascelle e all'inguine (segno di Crowe) si riscontrano più speso a 3-5 anni di età. Queste efelidi sono tipicamente piccole (2-3 mm di diametro), iperpigmentate.
Altre sedi delle efelidi sono: l'area fra le palpebre, intorno al collo, sotto il seno. In alcuni pazienti le efelidi possono essere presenti al di là di queste regioni.
Noduli di Lisch
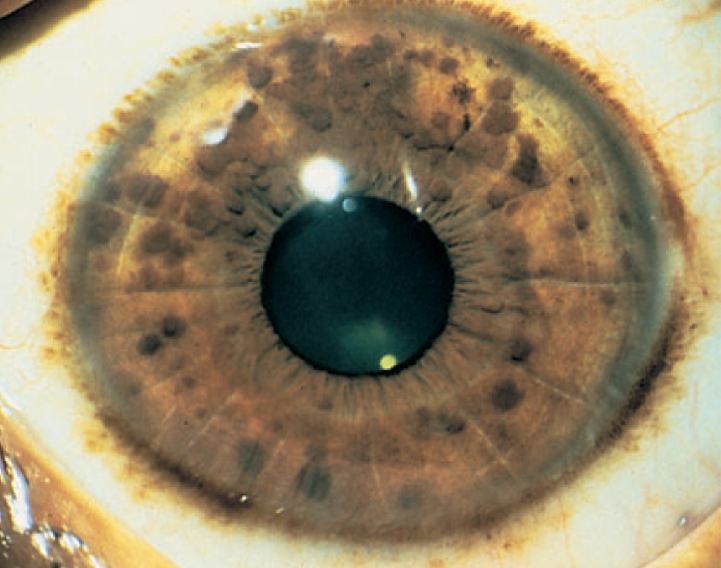
I noduli di Lisch sono degli amartomi melanocitici dell'iride che non disturbano la visione (Figura 3). Essi tipicamente compaiono fra i 5 e i 10 anni di età: si rendono ben visibili a un oculista con esperienza mediante una lampada a fessura. I noduli di Lisch sono patognomonici della NF1 e debbono essere distinti dai nevi dell'iride, che si osservano nella popolazione in generale.
Figura 3. Neurofibromatosi 1. Amartomi pigmentati dell'iride (noduli di Lisch) (Zitelli BJ et al, 2002)
Neurofibromi
I neurofibromi sono tumori delle cellule di Schwann che derivano dal tessuto fibroso che circonda la guaina dei nervi periferici; essi sono composti dalle cellule di Schwann, che costituiscono l'origine del tumore. A volte i pazienti presentano un prurito localizzato, associato ai neurofibromi, forse dovuto all'attivazione delle mast-cellen e alla loro successiva degranulazione. Non c'è accordo sull'uso degli antistaminici, mentre sono raccomandati gli emollienti per prevenire l'irritazione e viene raccomandato di evitare il caldo estremo.
I neurofibromi possono essere suddivisi in 4 gruppi:
Cutanei focali o diffusi
Sottocutanei
Plessiformi nodulari o diffusi
Spinali
La localizzazione e il numero di neurofibromi non è prevedibile, perché essi variano fra un paziente e l'altro anche nella stessa famiglia. I neurofibromi cutanei e sottocutanei insorgono tardivamente nella fanciullezza o precocemente nell'adolescenza, raramente causano dolore e non si trasformano in tumori maligni. Determinano sensazione di disagio nel paziente e un'estrema deturpazione dell'aspetto, per cui spesso richiedono l'intervento del chirurgo. I pazienti vanno avvertiti che possono comparire delle cicatrici ipertrofiche dopo l'operazione. Il laser a CO2 può essere utile per la rimozione delle lesioni più piccole.
La rimozione dei neurofibromi sottocutanei è più facile che esiti in deficit neurologici: questo tipo di neurofibromi si evidenzia alla palpazione della cute e può essere accompagnato da dolore o sensazione di prurito lungo il nervo colpito. I neurofibromi spinali possono insorgere in una o più radici nervose e possono associarsi a deficit neurologici sensitivi o motori.
Neurofibromi plessiformi
I neurofibromi plessiformi differiscono dai neurofibromi focali cutanei, perché originano da molti fasci nervosi e tendono a crescere lungo la lunghezza del nervo. Queste lesioni tipicamente sono presenti alla nascita e continuano a comparire fino alla tarda adolescenza e alla prima parte dell'età adulta. Essi insorgono in circa il 30% dei pazienti con neurofibromatosi, senza contare le lesioni interne che spesso rimangono non diagnosticate. Essi sono caratterizzati da un periodo di rapida crescita durante l'adolescenza, seguito da periodi di apparente inattività. I neurofibromi plessiformi ingrandiscono circondando strutture diverse, come la cute, la faccia, i muscoli, le ossa e gli organi interni, dando spesso dolore.
Si suddividono in 3 categorie:
Superficiali
Spostanti
Invasivi
Essi sono ben valutabili come grandezza con la risonanza magnetica. I neurofibromi plessiformi spostanti e invasivi sono i più pericolosi per la loro capacità di trasformasi in tumori maligni delle guaine dei nervi periferici.
I neurofibromi plessiformi possono essere trattati con la chirurgia, ma la loro caratteristica infiltrazione rende spesso incompleta l'escissione. La resezione di forme piccole e superficiali nel bambino può prevenire le complicazioni, associate a queste lesioni più tardi nella vita. Tuttavia va tenuto presente il rischio di associazione con deficit neurologici, associati alla rimozione del tumore.
Tumori maligni della guaina dei nervi periferici
I pazienti con neurofibromatosi hanno un rischio del 10% di sviluppare un sarcoma aggressivo a cellule fusiformi, indicato con l'acronimo MPNST. Questi tumori derivano da neurofibromi plessiformi, anche se nel 36% di questi tumori maligni non viene descritto un precedente neurofibroma plessiforme.
I pazienti con MPNST hanno in generale dolore e deficit neurologici. La tomografia a emissione di positroni con fluorodesossiglucosio è una prova sensibile e specifica per differenziare i neurofibromi plessiformi benigni dai MPNST. Gli MPNST sono spesso resistenti alla terapia e spesso metastatizzano, per cui hanno una cattiva prognosi. La cura standard consiste nell'escissione chirurgica seguita dalla radioterapia. L'uso della chemioterapia rimane controverso.
Neuropatia neurofibromatosa
È importante distinguere i deficit senso-motori, che avvengono come conseguenza di una neurofibromatosi spinale dalla neuropatia neurofibromatosa che colpisce l'1,3% dei pazienti con neurofibromatosi. Questa neuropatia è una rara manifestazione della neurofibromatosi: essa si presenta come una forma distale, simmetrica, caratterizzata clinicamente dallo sviluppo precoce di un gran numero di neurofibromi sottocutanei. In contrasto con la neuropatia associata alla neurofibromatosi tipo 2 (che comprende il 10% delle neurofibromatosi ed è caratterizzata da neurinomi bilaterali dell'acustico e da un genitore con una forma simile), la neuropatia della neurofibromatosi tipo 1 è accompagnata da un deficit sensoriale senza deterioramento clinico o neurofisiologico.
Displasia scheletrica
I pazienti con neurofibromatosi 1 presentano molte displasie scheletriche:
Bassa statura
Scoliosi distrofica
Pseudoartrosi tibiale (falsa articolazione che si forma in seguito a una frattura scomposta)
Displasia dell'ala dello sfenoide
Il 14% dei pazienti con neurofibromatosi si situa al di sotto delle 2 DS per l'altezza, in rapporto alla loro età. La scoliosi colpisce dal 10 al 26% dei pazienti, per cui i bambini vanno esaminati annualmente sotto questo riguardo: nei casi leggeri può bastare un busto, nei casi più gravi è necessaria una correzione chirurgica. La forma più grave (10% dei pazienti con neurofibromatosi) necessita di un intervento precoce per evitare compressioni midollari con paralisi degli arti e disfunzioni dell'intestino e della vescica urinaria.
La displasia dell'ala dello sfenoide comporta un difetto unilaterale del piatto orbitale e dell'osso frontale, che si può presentare come forma isolata senza altri sintomi.
La displasia delle ossa lunghe, come la displasia tibiale congenita con pseudoartrosi, porta a un'incurvatura della gamba. L'osso presenta un assottigliamento della corticale, che predispone a fratture patologiche per il carico del peso nel primo anno di vita. Le ripetute fratture e l'incapacità a guarire determina la pseudoartrosi. La pseudoartrosi risponde poco alla chirurgia per la grave osteopenia, per cui alcuni pazienti vanno sottoposti ad amputazione. Un trattamento precoce con bifosfonati può risultare utile.
L'osteopenia può portare a un'osteoporosi e a fratture più tardi nella vita. Avviare i bambini all'esercizio fisico può migliorare la situazione dell'osso.